Bloom syndrome
| Bloom syndrome | |
|---|---|
 | |
| Crystal structure of the Bloom's syndrome helicase BLM in complex with DNA (PDB ID: 4CGZ). | |
Bloom syndrome (often abbreviated as BS in literature)[1] is a rare autosomal recessive genetic disorder characterized by short stature, predisposition to the development of cancer, and genomic instability. BS is caused by mutations in the BLM gene which is a member of the RecQ DNA helicase family. Mutations in other members of this family, namely WRN and RECQL4, are associated with the clinical entities Werner syndrome and Rothmund–Thomson syndrome, respectively. More broadly, Bloom syndrome is a member of a class of clinical entities that are characterized by chromosomal instability, genomic instability, or both and by cancer predisposition.
Cells from a person with Bloom syndrome exhibit a striking genomic instability that includes excessive crossovers between homologous chromosomes and sister chromatid exchanges (SCEs). The condition was discovered and first described by New York dermatologist Dr. David Bloom in 1954.[2]
Bloom syndrome has also appeared in the older literature as Bloom–Torre–Machacek syndrome.[3] [4] The condition is named for Bloom, who described it in 1954.[4]
Signs and symptoms
The most prominent feature of Bloom syndrome is proportional small size. The small size is apparent in utero. At birth, neonates exhibit rostral to caudal lengths, head circumferences, and birth weights that are typically below the third percentile.[5]
The second most commonly noted feature is a rash on the face that develops early in life as a result of sun exposure. The facial rash appears most prominently on the cheeks, nose, and around the lips. It is described as erythematous, that is red and inflamed, and telangiectatic, that is characterized by dilated blood vessels at the skin's surface. The rash commonly also affects the backs of the hands and neck, and it can develop on any other sun-exposed areas of the skin. The rash is variably expressed, being present in a majority but not all persons with Bloom syndrome, and it is on average less severe in females than in males. Moreover, the sun sensitivity can resolve in adulthood. There are other dermatologic changes, including hypo-pigmented and hyper-pigmented areas, cafe-au-lait spots, and telangiectasias, which can appear on the face and on the ocular surface.[citation needed]
There is a characteristic facial appearance that includes a long, narrow face; prominent nose, cheeks, and ears; and micrognathism or undersized jaw. The voice is high-pitched and squeaky.[citation needed]
-
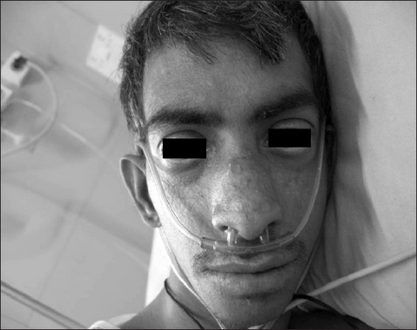
Telangiectasia over cheeks, prominent nose, dolicocephaly
-

An individual with Bloom syndrome


There are a variety of other features that are commonly associated with Bloom syndrome. There is a moderate immune deficiency, characterized by deficiency in certain immunoglobulin classes and a generalized proliferative defect of B and T cells. The immune deficiency is thought to be the cause of recurrent pneumonia and middle ear infections in persons with the syndrome.[6] Infants can exhibit frequent gastrointestinal upsets, with reflux, vomiting, and diarrhea, and there is a remarkable lack in interest in food. There are endocrine disturbances, particularly abnormalities of carbohydrate metabolism, insulin resistance and susceptibility to type 2 diabetes, dyslipidemia, and compensated hypothyroidism.[7] Persons with Bloom syndrome exhibit a paucity of subcutaneous fat. There is reduced fertility, characterized by a failure in males to produce sperm (azoospermia) and premature cessation of menses (premature menopause) in females. Despite these reductions, several women with Bloom syndrome have had children, and there is a single report of a male with Bloom syndrome bearing children.[8]
Although some persons with Bloom syndrome can struggle in school with subjects that require abstract thought, there is no evidence that intellectual disability is more common in Bloom syndrome than in other people.
The most serious and frequent complication of Bloom syndrome is cancer. In the 281 persons followed by the Bloom Syndrome Registry, 145 persons (51.6%) have been diagnosed with a malignant neoplasm, and there have been 227 malignancies.[9] The types of cancer and the anatomic sites at which they develop resemble the cancers that affect persons in the general population. The age of diagnosis for these cancers is earlier than for the same cancer in normal persons. And many persons with Bloom syndrome have been diagnosed with multiple cancers. The average life span is approximately 27 years. The most common cause of death in Bloom syndrome is from cancer. Other complications of the disorder include chronic obstructive lung disease and type 2 diabetes.[10]
There are a variety of excellent sources for more detailed clinical information about Bloom syndrome.[9]
There is a closely related entity that is now referred to as Bloom-syndrome-like disorder (BSLD) which is caused by mutations in components of the same protein complex to which the BLM gene product belongs, including TOP3A, which encodes the type I topoisomerase, topoisomerase 3 alpha, RMI1, and RMI2. The features of BSLD include small size and dermatologic findings, such as cafe-au-lait spots, and the presence of the once pathognomonic elevated SCEs is reported for persons with mutations in TOP3A and RMI1.[11][12]
Bloom syndrome shares some features with Fanconi anemia possibly because there is overlap in the function of the proteins mutated in this related disorder.[13]
Genetics


Bloom syndrome is an autosomal recessive disorder, caused by mutations in the maternally- and paternally-derived copies of the gene BLM.[14] As in other autosomal recessive conditions, the parents of an individual with Bloom syndrome do not necessarily exhibit any features of the syndrome. The mutations in BLM associated with Bloom syndrome are nulls and missense mutations that are catalytically inactive.[15] The cells from persons with Bloom syndrome exhibit a striking genomic instability that is characterized by hyper-recombination and hyper-mutation. Human BLM cells are sensitive to DNA damaging agents such as UV and methyl methanesulfonate,[16] indicating deficient repair capability. At the level of the chromosomes, the rate of sister chromatid exchange in Bloom's syndrome is approximately 10 fold higher than normal and quadriradial figures, which are the cytologic manifestations of crossing-over between homologous chromosome, are highly elevated. Other chromosome manifestations include chromatid breaks and gaps, telomere associations, and fragmented chromosomes.[17] The hyper-recombination can also be detected by molecular assays [18] The BLM gene is a member of the protein family referred to as RecQ helicases. The diffusion of BLM has been measured to 1.34 in nucleoplasm and 0.13 at nucleoli [19] DNA helicases are enzymes that attach to DNA and temporarily unravel the double helix of the DNA molecule. DNA helicases function in DNA replication and DNA repair. BLM very likely functions in DNA replication, as cells from persons with Bloom syndrome exhibit multiple defects in DNA replication, and they are sensitive to agents that obstruct DNA replication.[10]


The BLM helicase is a member of a protein complex with topoisomerase III alpha, RMI1 and RMI2, also known as BTRR or the dissolvasome.[20] Bloom-like phenotypes have been associated with mutations in topoisomerase III alpha, RMI1[21] and RMI2 genes.[11]
Relationship to cancer and aging
As noted above, there is greatly elevated rate of mutation in Bloom syndrome and the genomic instability is associated with a high risk of cancer in affected individuals.[22] The cancer predisposition is characterized by 1) broad spectrum, including leukemias, lymphomas, and carcinomas, 2) early age of onset relative to the same cancer in the general population, and 3) multiplicity, that is, synchronous or metachronous cancers. There is at least one person with Bloom syndrome who had five independent primary cancers. Persons with Bloom syndrome may develop cancer at any age. The average age of cancer diagnoses in the cohort is approximately 26 years old.[23]
Pathophysiology
When a cell prepares to divide to form two cells, the chromosomes are duplicated so that each new cell will get a complete set of chromosomes. The duplication process is called DNA replication. Errors made during DNA replication can lead to mutations. The BLM protein is important in maintaining the stability of the DNA during the replication process. Lack of BLM protein or protein activity leads to an increase in mutations; however, the molecular mechanism(s) by which BLM maintains stability of the chromosomes is still a very active area of research.[citation needed]
Persons with Bloom syndrome have an enormous increase in exchange events between homologous chromosomes or sister chromatids (the two DNA molecules that are produced by the DNA replication process); and there are increases in chromosome breakage and rearrangements compared to persons who do not have Bloom's syndrome. Direct connections between the molecular processes in which BLM operates and the chromosomes themselves are under investigation. The relationships between molecular defects in Bloom syndrome cells, the chromosome mutations that accumulate in somatic cells (the cells of the body), and the many clinical features seen in Bloom syndrome are also areas of intense research.[citation needed]
Diagnosis
Bloom syndrome is diagnosed using any of three tests - the presence of quadriradial (Qr, a four-armed chromatid interchange) in cultured blood lymphocytes, and/or the elevated levels of sister chromatid exchange in cells of any type, and/or the mutation in the BLM gene. The US Food and Drug Administration (FDA) announced on February 19, 2015 that they have authorized marketing of a direct-to-consumer genetic test from 23andMe.[24] The test is designed to identify healthy individuals who carry a gene that could cause Bloom Syndrome in their offspring.[9]
Treatment
Bloom syndrome has no specific treatment; however, avoiding sun exposure and using sunscreens can help prevent some of the cutaneous changes associated with photo-sensitivity. Efforts to minimize exposure to other known environmental mutagens are also advisable.[citation needed]
Epidemiology
Bloom syndrome is an extremely rare disorder in most populations and the frequency of the disease has not been measured in most populations. However, the disorder is relatively more common amongst people of Central and Eastern European Ashkenazi Jewish background. Approximately 1 in 48,000 Ashkenazi Jews are affected by Bloom syndrome, who account for about one-third of affected individuals worldwide.[25]
Bloom's Syndrome Registry
The Bloom's Syndrome Registry lists 265 individuals reported as suffering from this rare disorder (as of 2009), collected from the time it was first recognized in 1954. The registry was developed as a surveillance mechanism to observe the effects of cancer in the patients, which has shown 122[26] individuals have been diagnosed with cancer. It also acts as a report to show current findings and data on all aspects of the disorder.[27]
See also
References
- ↑ Online Mendelian Inheritance in Man (OMIM): Bloom Syndrome; BLM - 210900
- ↑ Bloom D (1954). "Congenital telangiectatic erythema resembling lupus erythematosus in dwarfs; probably a syndrome entity". American Journal of Diseases of Children. 88 (6): 754–8. doi:10.1001/archpedi.1954.02050100756008. PMID 13206391.
- ↑ James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology (10th ed.). Saunders. p. 575. ISBN 978-0-7216-2921-6.
- ↑ 4.0 4.1 Mulliken, John B. (2013). "13. Capillary malformations, hyperkeratotic stains, telangiectasias, and miscellaneous vascular blots". In Mulliken, John B.; Burrows, Patricia E.; Fishman, Steven J. (eds.). Mulliken and Young's Vascular Anomalies: Hemangiomas and Malformations (2nd ed.). Oxford University Press. p. 551. ISBN 978-0-19-972254-9. Archived from the original on 2023-06-30. Retrieved 2023-05-19.
- ↑ Keller, C; et al. (Apr 1999). "Growth deficiency and malnutrition in Bloom syndrome". J Pediatr. 134 (4): 472–479. doi:10.1016/s0022-3476(99)70206-4. PMID 10190923.
- ↑ German, James M.D. (November 1993). "Bloom Syndrome: A Mendelian Prototype of Somatic Mutational Disease". Medicine. 72 (6): 393–406. doi:10.1097/00005792-199311000-00003. PMID 8231788. S2CID 31448222.
- ↑ Diaz, A; et al. (Jun 9, 2006). "Evaluation of short stature, carbohydrate metabolism and other endocrinopathies in Bloom's syndrome". Horm Res. 66 (3): 111–117. doi:10.1159/000093826. PMID 16763388. S2CID 27176412.
- ↑ Ben Salah, G; et al. (Nov 2014). "A novel frameshift mutation in BLM gene associated with high sister chromatid exchanges (SCE) in heterozygous family members". Mol Biol Rep. 41 (11): 7373–7380. doi:10.1007/s11033-014-3624-5. PMID 25129257. S2CID 11074294.
- ↑ 9.0 9.1 9.2 Flanagan & Cunniff (2019).
- ↑ 10.0 10.1 Cunniff, Christopher; Bassetti, Jennifer A.; Ellis, Nathan A. (2017). "Bloom's Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition". Molecular Syndromology. 8 (1): 4–23. doi:10.1159/000452082. ISSN 1661-8769. PMC 5260600. PMID 28232778.
- ↑ 11.0 11.1 Hudson, Damien F.; et al. (15 December 2016). "Loss of RMI2 Increases Genome Instability and Causes a Bloom-Like Syndrome". PLOS Genet. 12 (12). e1006483. doi:10.1371/journal.pgen.1006483. PMC 5157948. PMID 27977684.
- ↑ Martin CA, et al. (2018). "Mutations in TOP3A Cause a Bloom Syndrome-like Disorder". Am J Hum Genet. 103 (2): 221–231. doi:10.1016/j.ajhg.2018.07.001. PMC 6080766. PMID 30057030.
- ↑ Deans AJ, West SC (December 2009). "FANCM connects the genome instability disorders Bloom syndrome and Fanconi Anemia". Mol. Cell. 36 (6): 943–53. doi:10.1016/j.molcel.2009.12.006. PMID 20064461.
- ↑ Ellis NA, Groden J, Ye TZ, Straughen J, Ciocci S, Lennon DJ, Proytcheva M, Alhadeff B, German J (1995). "The Bloom's syndrome gene product is homologous to RecQ helicases". Cell. 83 (4): 655–666. doi:10.1016/0092-8674(95)90105-1. PMID 7585968. S2CID 13439128.
- ↑ German J, Ciocci S, Ye TZ, Sanz MM, Ellis NA (2007). "Syndrome-causing mutations at BLM in persons in the Bloom's Syndrome Registry". Human Mutation. 28 (8): 743–753. doi:10.1002/humu.20501. PMID 17407155. S2CID 44382072.
- ↑ So S, Adachi N, Lieber MR, Koyama H (2004). "Genetic interactions between BLM and DNA ligase IV in human cells". J. Biol. Chem. 279 (53): 55433–42. doi:10.1074/jbc.M409827200. PMID 15509577.
- ↑ German J (Jan 1995). "Bloom's syndrome". Dermatol Clin. 13 (1): 7–18. doi:10.1016/S0733-8635(18)30101-3. PMID 7712653.
- ↑ Langlois RG, Bigbee WL, Jensen RH, German J (Jan 1989). "Evidence for increased in vivo mutation and somatic recombination in Bloom's syndrome". Proc Natl Acad Sci U S A. 86 (2): 670–4. Bibcode:1989PNAS...86..670L. doi:10.1073/pnas.86.2.670. PMC 286535. PMID 2911598.
- ↑ Kristian Moss Bendtsen; Martin Borch Jensen; Alfred May; Lene Juel Rasmussen; Ala Trusina; Vilhelm A. Bohr; Mogens H. Jensen (2014). "Dynamics of the DNA repair proteins WRN and BLM in the nucleoplasm and nucleoli". European Biophysics Journal. 43 (10–11): 509–16. doi:10.1007/s00249-014-0981-x. PMC 5576897. PMID 25119658.
- ↑ Bizard, A. H.; Hickson, I. D. (1 July 2014). "The Dissolution of Double Holliday Junctions". Cold Spring Harbor Perspectives in Biology. 6 (7): a016477. doi:10.1101/cshperspect.a016477. PMC 4067992. PMID 24984776.
- ↑ Martin, Carol-Anne; et al. (August 2018). "Mutations in TOP3A Cause a Bloom Syndrome-like Disorder". The American Journal of Human Genetics. 103 (2): 221–231. doi:10.1016/j.ajhg.2018.07.001. PMC 6080766. PMID 30057030.
- ↑ German J (Jan 1997). "Bloom's syndrome. XX. The first 100 cancers". Cancer Genet Cytogenet. 93 (1): 100–6. doi:10.1016/s0165-4608(96)00336-6. PMID 9062585.
- ↑ "Bloom Syndrome Registry | Pediatrics". Archived from the original on 2018-05-09. Retrieved 2021-10-23.
- ↑ "FDA permits marketing of first direct-to-consumer genetic carrier test for Bloom syndrome". U.S. Food and Drug Administration. Archived from the original on 3 May 2015. Retrieved 19 May 2015.
- ↑ Li L, Eng C, Desnick B, German J, Ellis NA (1998). "Carrier frequency of the Bloom syndrome blmAsh mutation in the Ashkenazi Jewish population". Mol Genet Metab. 64 (4): 286–290. doi:10.1006/mgme.1998.2733. PMID 9758720.
- ↑ "Data from the Bloom's Syndrome Registry, 2009". Weill Cornell Medical College. Weill Cornell Medical Center. 2009. Archived from the original on 4 June 2018. Retrieved 17 April 2015.
- ↑ German, James; Bloom, David; Passarge, Eberhard (23 April 2008). "Bloom's syndrome. V. Surveillance for cancer in affected families". Clinical Genetics. 12 (3): 162–168. doi:10.1111/j.1399-0004.1977.tb00919.x. PMID 908169. S2CID 40914579.
- Flanagan M, Cunniff CM (February 14, 2019) [March 22, 2006]. Adam MP, Ardinger HH, Pagon RA, et al. (eds.). "Bloom syndrome". GeneReviews. Seattle (WA): University of Washington. PMID 20301572. Archived from the original on September 16, 2013. Retrieved July 14, 2019.
Further reading
- Margaret P Adam; Holly H Ardinger; Roberta A Pagon; Stephanie E Wallace; et al., eds. (1993). GeneReviews. Seattle (WA): University of Washington. ISSN 2372-0697. Archived from the original on 2019-09-10. Retrieved 2021-10-23.
External links
| Classification | |
|---|---|
| External resources |
- Bloom syndrome at NLM Genetics Home Reference
- Pages with script errors
- All articles with unsourced statements
- Articles with unsourced statements from November 2020
- Articles with invalid date parameter in template
- Articles with unsourced statements from September 2021
- Chromosome instability syndromes
- Genodermatoses
- Autosomal recessive disorders
- Rare syndromes
- Ashkenazi Jews topics
- IUIS-PID table 3 immunodeficiencies
- DNA replication and repair-deficiency disorders
- Progeroid syndromes
- Syndromes with tumors
- Syndromes affecting stature