Pachyonychia congenita
| Pachyonychia congenita | |
|---|---|
.jpg) | |
| Pachyonychia congenita | |
| Specialty | Dermatology |
Pachyonychia congenita (PC) is a rare group of autosomal dominant skin disorders that are caused by a mutation in one of five different keratin genes.
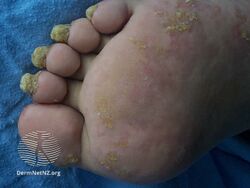
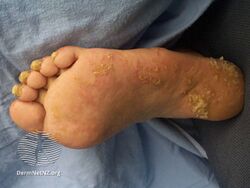
Pachyonychia congenita is often associated with thickened toenails, plantar keratoderma, and plantar pain.[1]
Signs and symptoms
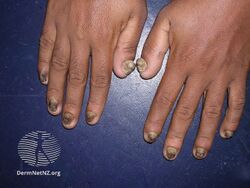
Pachyonychia congenita is characterized by a clinical triad present in 97% of people with PC by the time they turn 10 years old:[2][3]
- Thickened toenails
- Plantar keratoderma
- Plantar pain that may require some patients to use wheelchairs, canes, crutches, and pain medications due to its severity
Other signs and symptoms found in PC include:[2][4]
- Thickened fingernails
- Palmar keratoderma
- Oral leukokeratosis
- Cysts, including steatocystoma multiplex
- Follicular hyperkeratosis
- Natal or prenatal teeth
- Blisters
- Excessive sweating of the palms and soles
- Excess earwax production
- Ear pain
- Hoarseness
- Angular chelitis
- Fingernail and toenail infections
-
Pachyonychia congenita
-
Pachyonychia congenita
-
Pachyonychia congenita
.jpg)
.jpg)
.jpg)
Cause

The condition is caused by genetic mutations in one of five genes that encode keratin proteins. Three keratin genes were identified to have a role PC in 1995[5][6] with a fourth keratin gene's role in PC identified in 1998.[7]
Inheritance
Pachyonychia congenita follows an autosomal dominant pattern of inheritance, which means the defective gene is located on an autosome, and only one copy of the gene is required to inherit the disorder from a parent who has the disorder. On average, 50% of the offspring of an affected person will inherit the disorder, regardless of sex.
Occasionally, however, a solitary case can emerge in a family with no prior history of the disorder due to the occurrence of a new mutation (often referred to as a sporadic, spontaneous or de novo mutation).
Diagnosis
Classification
ILDS: Q84.520 ICD-10: Q84.5
Pachyonychia congenita consists of five sub-types, each named after its corresponding genetic mutation and each associated with distinguishing clinical features:[2][8]
- PC-K6a is caused by a mutation in the KRT6A gene and more often associated with oral leukokeratosis and poor feeding in infants.
- PC-K6b is caused by a mutation in the KRT6B gene and more commonly associated with an increased age of onset (>14 years).
- PC-K6c is caused by a mutation in the KRT6C gene and is the least common sub-type. It is not often associated with the additional features of oral leukokeratosis, cysts, follicular hyperkeratosis, and natal teeth.
- PC-K16 is caused by a mutation in the KRT16 gene and is more commonly associated with severe plantar pain.
- PC-K17 is caused by a mutation in the KRT17 gene and more commonly associated with the presence of cysts, follicular kyperkeratosis, and natal teeth.
Before the genetic basis of Pachyonychia congenita was identified and described, the disease was historically divided into the following sub-types:[9]: 510
- Pachyonychia congenita type I (also known as "Jadassohn–Lewandowsky syndrome"[10]) is an autosomal dominant keratoderma that principally involves the plantar surfaces, but also with nails changes that may be evident at birth, but more commonly develop within the first few months of life.[9]: 510 [10][11]: 569
- Pachyonychia congenita type II (also known as "Jackson–Lawler pachyonychia congenita" and "Jackson–Sertoli syndrome") is an autosomal dominant keratoderma presenting with a limited focal plantar keratoderma that may be very minor, with nails changes that may be evident at birth, but more commonly develop within the first few months of life.[9][11]: 569
Clinical Diagnosis
In order to clinically diagnose pachyonychia congenita, the clinical triad of toenail thickening, plantar keratoderma, and plantar pain must be present. This triad is present in 97% of individuals with PC by the age of 10 years old.[2]
Pachyonychia congenita can be suspected in patients who do not have the complete clinical triad but who exhibit other symptoms such as cysts, oral leukokeratosis, follicular hyperkeratosis, palmoplantar hyperhidrosis, or natal teeth. Since PC is inherited in an autosomal dominant fashion in 70% of individuals, it should especially be suspected in patients with symptoms who also have a parent with similar symptoms. Histopathological analysis of skin or nail tissue is not helpful in diagnosis of PC, but can be used to rule out some related diseases. If there is a clinical suspicion for PC, genetic testing can confirm the diagnosis.[2]
Genetic Diagnosis
The diagnosis of PC can be confirmed by the identification of a mutation in one of the five genes responsible for the condition: KRT6A, KRT6B, KRT6C, KRT16, KRT17. Pachyonychia Congenita Project is a non-profit dedicated to finding a cure for PC. The organization houses a genetic registry (the International PC Research Registry) and offers free genetic testing for individuals suspected to have PC.[12]
Treatment
There is currently no cure for pachyonychia congenita. Treatment focuses on symptom relief for any plantar pain, hyperkeratoses, cysts, leukokeratosis, hyperhidrosis, or secondary infections.[13]
Palmoplantar keratoderma can be treated with consistent grooming, including trimming back the callus, applying emollients, and draining blisters. Plantar pain is often treated by reducing pressure on the feet by minimizing walking, wearing cushioned footwear, or using wheelchairs or crutches. Hyperkeratosis can be treated with keratolytic emollients while cysts may be treated with incision and drainage. Patients with hyperhidrosis may need to wear moisture-wicking socks and ventilated shoes. Any secondary infection may need to be treated with antibiotics, though infection can often be prevented with appropriate grooming and vinegar or bleach baths.[13][2][8]
Epidemiology
Pachyonychia congenita is a rare disorder with an unknown prevalence. As of 2018, the International PC Research Registry has identified approximately 774 individuals with the disease, but prevalence is estimated to be 5,000–10,000 worldwide.[2][8] The disease affects both males and females.
Research
There are several ongoing investigational therapies for pachyonychia congenita, including topical sirolimus, siRNA, botulinum toxin, statins, and anti-TNF biologics.[2] Pachyonychia Congenita Project houses a list of clinical trials and assists with clinical trial recruitment from patients enrolled in their International PC Research Registry.[12][14]
See also
References
- ↑ Bellet, Jane Sanders (2021). "Paediatric nail disorders". In Lipner, Shari (ed.). Nail Disorders: Diagnosis and Management, An Issue of Dermatologic Clinics. Philadelphia: Elsevier. p. 233. ISBN 978-0-323-70923-1.
- ↑ 2.0 2.1 2.2 2.3 2.4 2.5 2.6 2.7 Smith, Frances JD; Hansen, C. David; Hull, Peter R.; Kaspar, Roger L.; McLean, WH Irwin; O’Toole, Edel; Sprecher, Eli (1993), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "Pachyonychia Congenita", GeneReviews®, University of Washington, Seattle, PMID 20301457, archived from the original on 2019-08-01, retrieved 2018-08-01
- ↑ Eliason, Mark J.; Leachman, Sancy A.; Feng, Bing-jian; Schwartz, Mary E.; Hansen, C. David (October 2012). "A review of the clinical phenotype of 254 patients with genetically confirmed pachyonychia congenita". Journal of the American Academy of Dermatology. 67 (4): 680–686. doi:10.1016/j.jaad.2011.12.009. ISSN 1097-6787. PMID 22264670.
- ↑ "What Is Pachyonychia Congenita?". www.pachyonychia.org. Archived from the original on 2018-08-02. Retrieved 2018-08-01.
- ↑ McLean W, Rugg E, Lunny D, Morley S, Lane E, Swensson O, Dopping-Hepenstal P, Griffiths W, Eady R, Higgins C, Navsaria H, Leigh I, Strachan T, Kunkeler L, Munro C (1995). "Keratin 16 and keratin 17 mutations cause pachyonychia congenita". Nature Genetics. 9 (3): 273–8. doi:10.1038/ng0395-273. PMID 7539673. S2CID 1873772.
- ↑ Bowden PE, Haley JL, Kansky A, Rothnagel JA, Jones DO, Turner RJ (1995). "Mutation of a type II keratin gene (K6a) in pachyonychia congenita". Nature Genetics. 10 (3): 363–5. doi:10.1038/ng0795-363. PMID 7545493. S2CID 26060130.
- ↑ Smith FJ, Jonkman MF, Van Goor H, Coleman CM, Covello SP, Uitto J, McLean WH (1998). "A Mutation in Human Keratin K6b Produces a Phenocopy of the K17 Disorder Pachyonychia Congenita Type 2". Human Molecular Genetics. 7 (7): 1143–8. doi:10.1093/hmg/7.7.1143. PMID 9618173.
- ↑ 8.0 8.1 8.2 "Pachyonychia Congenita - NORD (National Organization for Rare Disorders)". NORD (National Organization for Rare Disorders). Archived from the original on 2018-08-02. Retrieved 2018-08-02.
- ↑ 9.0 9.1 9.2 Freedberg, et al. (2003). Fitzpatrick's Dermatology in General Medicine. (6th ed.). McGraw-Hill. ISBN 0-07-138076-0.
- ↑ 10.0 10.1 Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. p. 740. ISBN 978-1-4160-2999-1.
- ↑ 11.0 11.1 James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. ISBN 0-7216-2921-0.
- ↑ 12.0 12.1 "International PC Research Registry (IPCRR)". www.pachyonychia.org. Archived from the original on 2018-08-02. Retrieved 2018-08-01.
- ↑ 13.0 13.1 Goldberg, I.; Fruchter, D.; Meilick, A.; Schwartz, M.E.; Sprecher, E. (2013-01-30). "Best treatment practices for pachyonychia congenita". Journal of the European Academy of Dermatology and Venereology. 28 (3): 279–285. doi:10.1111/jdv.12098. ISSN 0926-9959. PMID 23363249. S2CID 29684041.
- ↑ "Clinical Trials & Studies". www.pachyonychia.org. Archived from the original on 2018-08-02. Retrieved 2018-08-01.
External links
| Classification | |
|---|---|
| External resources |
- GeneReviews/NCBI/NIH/UW entry on Pachyonychia Congenita Archived 2010-04-11 at the Wayback Machine
- OMIM: 260130 Pachyonychia congenita recessive at NIH's Office of Rare Diseases