Hairy cell leukemia
| Hairy cell leukemia | |
|---|---|
 | |
| Specialty | Hematology and oncology |
Hairy cell leukemia is an uncommon hematological malignancy characterized by an accumulation of abnormal B lymphocytes.[1] It is usually classified as a subtype of chronic lymphocytic leukemia (CLL). Hairy cell leukemia makes up about 2% of all leukemias, with fewer than 2,000 new cases diagnosed annually in North America and Western Europe combined.
Hairy cell leukemia (HCL) was originally described as histiocytic leukemia, malignant reticulosis, or lymphoid myelofibrosis in publications dating back to the 1920s. The disease was formally named leukemic reticuloendotheliosis, and its characterization was significantly advanced by Bertha Bouroncle and colleagues at the Ohio State University College of Medicine in 1958. Its common name, which was coined in 1966,[2] is derived from the "hairy" appearance of the malignant B cells under a microscope.[3]
Signs and symptoms
In HCL, the "hairy cells" (malignant B lymphocytes) accumulate in the bone marrow, interfering with the production of normal white blood cells, red blood cells, and platelets. Consequently, patients may develop infections related to low white blood cell count, anemia and fatigue due to a lack of red blood cells, or easy bleeding due to a low platelet count.[4] Leukemic cells may gather in the spleen and cause it to swell; this can have the side effect of making the person feel full even when he or she has not eaten much.[citation needed]
Hairy cell leukemia is commonly diagnosed after a routine blood count shows unexpectedly low numbers of one or more kinds of normal blood cells, or after unexplained bruises or recurrent infections in an otherwise apparently healthy patient.[citation needed]
Platelet function may be somewhat impaired in HCL patients, although this does not appear to have any significant practical effect.[5] It may result in somewhat more mild bruises than would otherwise be expected for a given platelet count or a mildly increased bleeding time for a minor cut, likely the result of producing slightly abnormal platelets in the overstressed bone marrow tissue.
Patients with a high tumor burden may also have somewhat reduced levels of cholesterol,[6] especially in patients with an enlarged spleen.[7] Cholesterol levels return to more normal values with successful treatment of HCL.
Cause
As with many cancers, the cause of HCL is unknown. Exposure to tobacco smoke, ionizing radiation, or industrial chemicals (with the possible exception of diesel) does not appear to increase the risk of developing it.[8] Farming and gardening correlate with an increased risk of HCL development in some studies which does not necessarily imply causation.[9]
A 2011 study identified somatic BRAF V600E mutations in all 47 HCL patients studied, and no such mutations in the 193 peripheral B-cell lymphomas/leukemias other than HCL.[10]
The U.S. Institute of Medicine (IOM) found a correlation which permits an association between exposure to herbicides and later development of chronic B-cell leukemias and lymphomas in general. The IOM report emphasizes that neither animal nor human studies indicate an association of herbicides with HCL specifically. However, the IOM extrapolated data from chronic lymphocytic leukemia and non-Hodgkin lymphoma to conclude that HCL and other rare B-cell neoplasms may share this risk factor.[11] As a result of the IOM report, the U.S. Department of Veterans Affairs considers HCL an illness presumed to be a service-related disability[citation needed]
Human T-lymphotropic virus 2 (HTLV-2) has been isolated in a small number of patients with the variant form of HCL.[12] In the 1980s, HTLV-2 was identified in a patient with a T-cell lymphoproliferative disease; this patient later developed HCL, but HTLV-2 was not found in the hairy cell clones.[13] There is no evidence that HTLV-II causes any sort of hematological malignancy, including HCL.[14]
Pathophysiology
Pancytopenia in HCL is caused primarily by marrow failure and splenomegaly. Bone-marrow failure is caused by the accumulation of hairy cells and reticulin fibrosis in the bone marrow, as well as by the detrimental effects of dysregulated cytokine production.[15] Splenomegaly reduces blood counts through sequestration, marginalization, and destruction of healthy blood cells inside the spleen.[15]
Hairy cells are nearly mature B cells, which are activated clonal cells with signs of VH gene differentiation.[16] They may be related to pre-plasma marginal zone B cells[15] or memory cells.
Cytokine production is disturbed in HCL. Hairy cells produce and thrive on TNF-alpha.[15] This cytokine also suppresses normal production of healthy blood cells in the bone marrow.[15]
Unlike healthy B cells, hairy cells express and secrete an immune system protein called interleukin-2 receptor (IL-2R).[15] In HCL-V, only part of this receptor is expressed.[15] As a result, disease status can be monitored by measuring changes in the amount of IL-2R in the blood serum.[15] The level increases as hairy cells proliferate, and decreases when they are killed. Although uncommonly used in North America and Northern Europe, this test correlates better with disease status and predicts relapse more accurately than any other test.
Hairy cells respond to normal production of some cytokines by T cells with increased growth. Treatment with interferon-alpha suppresses the production of this pro-growth cytokine from T cells.[15] A low level of T cells, which is commonly seen after treatment with cladribine or pentostatin, and the consequent reduction of these cytokines, is also associated with reduced levels of hairy cells.
In June 2011, E Tiacci et al[17][18] discovered that 100% of HCL samples analysed had the oncogenic BRAF mutation V600E, and proposed that this is the disease's driver mutation. Until this point, only a few genomic imbalances had been found in the hairy cells, such as trisomy 5 had been found.[15] The expression of genes is also dysregulated in a complex and specific pattern. The cells underexpress 3p24, 3p21, 3q13.3-q22, 4p16, 11q23, 14q22-q24, 15q21-q22, 15q24-q25, and 17q22-q24 and overexpress 13q31 and Xq13.3-q21.[19] It has not yet been demonstrated that any of these changes have any practical significance to the patient.
Diagnosis
The diagnosis of HCL may be suggested by abnormal results on a complete blood count (CBC), but additional testing is necessary to confirm the diagnosis. A CBC normally shows low counts for white blood cells, red blood cells, and platelets in HCL patients. However, if large numbers of hairy cells are in the blood stream, then normal or even high lymphocyte counts may be found.
On physical examination, 80–90% of patients have an enlarged spleen, which can be massive.[15] This is less likely among patients who are diagnosed at an early stage. Peripheral lymphadenopathy (enlarged lymph nodes) is uncommon (less than 5% of patients), but abdominal lymphadenopathy is a relatively common finding on computed tomography scans.[15]
-
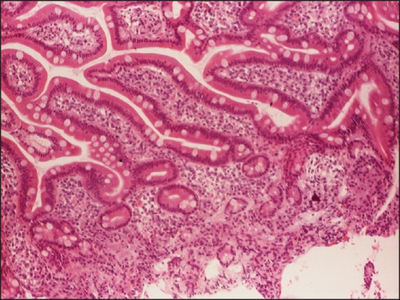
Numerous hairy cells admixed with other elements
-
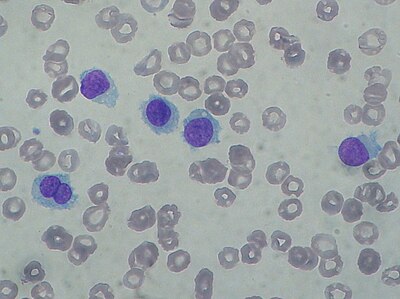
Hairy cell leukemia: Abnormal B cells look "hairy" under a microscope because of radial projections from their surface.


The most important laboratory finding is the presence of hairy cells in the bloodstream.[15] Hairy cells are abnormal white blood cells with hair-like projections of cytoplasm; they can be seen by examining a blood smear or bone marrow biopsy specimen. The blood film examination is done by staining the blood cells with Wright's stain and looking at them under a microscope. Hairy cells are visible in this test in about 85% of cases.[15]
Most patients require a bone-marrow biopsy for final diagnosis. The bone marrow biopsy is used both to confirm the presence of HCL and also the absence of any additional diseases, such as Splenic marginal zone lymphoma or B-cell prolymphocytic leukemia. The diagnosis can be confirmed by viewing the cells with a special stain known as TRAP (tartrate resistant acid phosphatase). More recently, DB44 testing assures more accurate results.
Definitively diagnosing HCL is also possible through flow cytometry on blood or bone marrow. The hairy cells are larger than normal and positive for CD19, CD20, CD22, CD11c, CD25, CD103, and FMC7 antigens.[20] (CD103, CD22, and CD11c are strongly expressed.)[21]
Hairy cell leukemia-variant (HCL-V), which shares some characteristics with B cell prolymphocytic leukemia (B-PLL), does not show CD25 (also called the interleukin-2 receptor, alpha).
The differential diagnoses include several kinds of anemia, including myelophthisis and aplastic anemia,[22] and most kinds of blood neoplasms, including hypoplastic myelodysplastic syndrome, atypical chronic lymphocytic leukemia, B-cell prolymphocytic leukemia, or idiopathic myelofibrosis.[16]
Classification
When not further specified, the "classic" form is often implied, but two variants have been described: Hairy cell leukemia-variant[23] and a Japanese variant.[citation needed] The non-Japanese variant is more difficult to treat than either 'classic' HCL or the Japanese HCL.[citation needed]
HCL-V
Hairy cell leukemia-variant (HCL-V) is usually described as a prolymphocytic variant of HCL.[24] It was first formally described in 1980 by a paper from the University of Cambridge's Hayhoe laboratory.[25] About 10% of people with HCL have this variant form of the disease, representing about 60-75 new cases of HCL-V each year in the U.S. While classic HCL primarily affects men, HCL-V is more evenly divided between males and females.[26] While the disease can appear at any age, the median age at diagnosis is over 70.[27]
Similar to B-cell prolymphocytic leukemia (B-PLL) in chronic lymphocytic leukemia, HCL-V is a more aggressive disease. Historically, it has been considered less likely to be treated successfully than is classic HCL, and remissions have tended to be shorter. The introduction of combination therapy with concurrent rituximab and cladribine therapy, though, has shown excellent results in early follow-up.[28] As of 2016, this therapy is considered the first-line treatment of choice for many people with HCL-V.[29]
Many older treatment approaches, such as interferon-alpha, the combination chemotherapy regimen "CHOP", and common alkylating agents such as cyclophosphamide, showed very little benefit.[26] Pentostatin and cladribine administered as monotherapy (without concurrent rituximab) provide some benefit to many people with HCL-V, but typically induce shorter remission periods and lower response rates than when they are used in classic HCL. More than half of people respond partially to splenectomy.[26]
In terms of B-cell development, the prolymphocytes are less developed than are lymphocytes or plasma cells, but are still more mature than their lymphoblastic precursors.
HCL-V differs from classic HCL principally in these respects:
- Higher white blood cell counts, sometimes exceeding 100,000 cells per microliter
- A more aggressive course of disease requiring more frequent treatment
- Hairy cells with an unusually large nucleolus for their size
- Production of little excess fibronectin produced by classic hairy cells[30] to interfere with bone marrow biopsies
- Low or no cell-surface expression of CD25 (interleukin-2 [IL-2] receptor alpha chain, or p55).[31]
Low levels of CD25, a part of the receptor for a key immunoregulating hormone, may explain why HCL-V cases are generally much more resistant to treatment by immune-system hormones.[24]
HCL-V, which usually features a high proportion of hairy cells without a functional p53 tumor suppressor gene, is somewhat more likely to transform into a higher-grade malignancy. A typical transformation rate of 5-6% has been postulated in the UK, similar to the Richter's transformation rate for SLVL and CLL.[26][32] Among HCL-V patients, the most aggressive cases normally have the least amount of p53 gene activity.[33] Hairy cells without the p53 gene tend, over time, to displace the less aggressive p53(+) hairy cells.
Some evidence suggests that a rearrangement of the immunoglobulin gene VH4-34, which is found in about 40% of HCL-V patients and 10% of classic HCL patients, may be a more important poor prognostic factor than variant status, with HCL-V patients without the VH4-34 rearrangement responding about as well as classic HCL patients.[34]
Hairy cell leukemia-Japanese variant
The variant called hairy cell leukemia-Japanese variant, or HCL-J, is more easily treated. Treatment with cladribine has been reported.[35]
Prevention
Because the cause is unknown, no effective preventive measures can be taken, but the U.S. Institute of Medicine permits an association to exposure to herbicides (atrazine). The U.S. Department of Veterans Affairs also considers it a service disability related to Agent Orange. Because the disease is rare, routine screening is not cost effective.[citation needed]
Treatment
Several treatments are available, and successful control of the disease is common.
- Not everyone needs treatment immediately. Treatment is usually given when the symptoms of the disease interfere with the patient's everyday life, or when white blood cell or platelet counts decline to dangerously low levels, such as an absolute neutrophil count below 1000 cells per microliter (1.0 K/μL). Not all patients need treatment immediately upon diagnosis.
- Treatment delays are less important than in solid tumors. Unlike most cancers, treatment success does not depend on treating the disease at an early stage. Because delays do not affect treatment success, no standards exist for how quickly a patient should receive treatment. Waiting too long, though, can cause its own problems, such as an infection that might have been avoided by proper treatment to restore immune-system function. Also, having a higher number of hairy cells at the time of treatment can make certain side effects somewhat worse, as some side effects are primarily caused by the body's natural response to the dying hairy cells. This can result in the hospitalization of a patient whose treatment would otherwise be carried out entirely at the hematologist's office.
- Single-drug treatment is typical. Unlike most cancers, only one drug is normally given to a patient at a time. While monotherapy is normal, combination therapy—typically using one first-line therapy and one second-line therapy—is being studied in current clinical trials, and is used more frequently for refractory cases. Combining rituximab with cladribine or pentostatin may or may not produce any practical benefit to the patient.[36] Combination therapy is almost never used with a new patient. Because the success rates with purine analog monotherapy are already so high, the additional benefit from immediate treatment with a second drug in a treatment-naïve patient is assumed to be very low. For example, one round of either cladribine or pentostatin gives the median first-time patient a decade-long remission; the addition of rituximab, which gives the median patient only three or four years, might provide no additional value for this easily treated patient. In a more difficult case, however, the benefit from the first drug may be substantially reduced, so a combination may provide some benefit.
First-line therapy
Cladribine (2CDA) and pentostatin (DCF) are the two most common first-line therapies. They both belong to a class of medications called purine analogs, which have mild side effects compared to traditional chemotherapy regimens.
Cladribine can be administered by injection under the skin, by infusion over a few hours into a vein (inravenous - IV), or by a pump worn by the patient that provides a slow drip into a vein, 24 hours a day for 7 days. Most patients receive cladribine by IV infusion once a day for five to seven days, but more patients are being given the option of taking this drug once a week for six weeks. The different dosing schedules used with cladribine are roughly equally effective and safe.[37] Relatively few patients have significant side effects other than fatigue and a high fever caused by the cancer cells dying, although complications such as infection and acute kidney failure have been seen. Pentostatin is chemically similar to cladribine, and has a similar success rate and side effect profile, but it is always given over a much longer period of time, usually one dose by IV infusion every two weeks for 3-6 months.
During the weeks following treatment, the patients' immune systems are severely weakened, but their bone marrow will begin to produce normal blood cells again. Treatment often results in long-term remission. About 85% of patients achieve a complete response from treatment with either cladribine or pentostatin, and another 10% receive some benefit from these drugs, although no permanent cure for this disease is known. If the cancer cells return, the treatment may be repeated and should again result in remission, although the odds of success decline with repeated treatment.[38] Remission lengths vary significantly, from one year to more than twenty years. The median patient can expect a treatment-free interval of about ten years.
2-Chlorodeoxyadenosine (Cladribine) induced complete responses in patients with hairy cell leukemia resistant to DCF , suggesting a lack of cross-resistance. Also, 2-CdA is not prohibitively toxic in patients intolerant of DCF (Pentostatin).[39]
Second-line therapy
If a patient is resistant to either cladribine or pentostatin, then second-line therapy is pursued.
- Monoclonal antibodies: The most common treatment for cladribine-resistant disease is infusing monoclonal antibodies that destroy cancerous B cells. Rituximab is by far the most commonly used. Most patients receive one IV infusion over several hours each week for 4-8 weeks. Two partial and 10 complete responses resulted fromf 15 patients with relapsed disease, for a total of 80% responding.[40] The median patient (including nonresponders) did not require further treatment for more than 3 years. This eight-dose study had a higher response rate than a four-dose study at Scripps, which achieved only 25% response rate.[41] Rituximab has successfully induced a complete response in Hairy Cell-Variant.[42]
Rituximab's major side effect is serum sickness, commonly described as an "allergic reaction", which can be severe, especially on the first infusion. Serum sickness is primarily caused by the antibodies clumping during infusion and triggering the complement cascade. Although most patients find that side effects are adequately controlled by antiallergy drugs, some severe, and even fatal, reactions have occurred. Consequently, the first dose is always given in a hospital setting, although subsequent infusions may be given in a physician's office. Remissions are usually shorter than with the preferred first-line drugs, but hematologic remissions of several years' duration are not uncommon. Other B cell-destroying monoclonal antibodies such as alemtuzumab, ibritumomab tiuxetan and I-131 tositumomab may be considered for refractory cases.
- Interferon-alpha is an immune system hormone that is very helpful to a relatively small number of patients, and somewhat helpful to most patients. In about 65% of patients,[43] the drug helps stabilize the disease or produce a slow, minor improvement for a partial response.[44]
The typical dosing schedule injects at least 3 million units of interferon-alpha (not pegylated versions) three times a week, although the original protocol began with 6 months of daily injections.
Some patients tolerate IFN-alpha very well after the first fw weeks, while others find that its characteristic flu-like symptoms persist. About 10% of patients develop a level of depression. By maintaining a steadier level of the hormone in the body, daily injections might cause fewer side effects in selected patients. Drinking at least 2 liters of water each day, while avoiding caffeine and alcohol, can reduce many of the side effects.
A drop in blood counts is usually seen during the first 1-2 months of treatment. Most patients find that their blood counts get worse for a few weeks immediately after starting treatment, although some patients find their blood counts begin to improve within just 2 weeks.[45]
Typically, 6 months are neededto figure out whether this therapy is useful. Common criteria for treatment success include:
- Normalization of hemoglobin levels (above 12.0 g/dL),
- A normal or somewhat low platelet count (above 100 K/µL), and
- A normal or somewhat low absolute neutrophil count (above 1.5 K/µL).[45]
If it is well tolerated, patients usually take the hormone for 12 to 18 months. An attempt may be made then to end the treatment, but most patients discover that they need to continue taking the drug for it to be successful. These patients often continue taking this drug indefinitely, until either the disease becomes resistant to this hormone, or the body produces an immune system response that limits the drug's ability to function. A few patients are able to achieve a sustained clinical remission after taking this drug for six months to one year. This may be more likely when IFN-alpha has been initiated shortly after another therapy. IFN-alpha is considered the drug of choice for pregnant women with active HCL, although it carries some risks, such as the potential for decreased blood flow to the placenta.
IFN-alpha works by sensitizing the hairy cells to the killing effect of the immune-system hormone TNF-alpha, whose production it promotes.[46] IFN-alpha works best on classic hairy cells that are not protectively adhered to vitronectin or fibronectin, which suggests that patients who encounter less fibrous tissue in their bone-marrow biopsies may be more likely to respond to IFN-alpha therapy. It also explains why unadhered hairy cells, such as those in the bloodstream, disappear during IFN-alpha treatment well before reductions are seen in adhered hairy cells, such as those in the bone marrow and spleen.[46]
Other treatments
Splenectomy can produce long-term remissions in patients whose spleens seem to be heavily involved, but its success rate is noticeably lower than cladribine or pentostatin. Splenectomies are also performed for patients whose persistently enlarged spleens cause significant discomfort or in patients whose persistently low platelet counts suggest idiopathic thrombocytopenic purpura.
Bone marrow transplants are usually shunned in this highly treatable disease because of the inherent risks in the procedure. They may be considered for refractory cases in younger, otherwise healthy individuals. "Minitransplants" are possible.
People with low numbers of red blood cells or platelets may also receive red blood cells and platelets through blood transfusions. Blood transfusions are always irradiated to remove white blood cells and thereby reduce the risk of graft-versus-host disease. Affected people may also receive a hormone to stimulate production of red blood cells. These treatments may be medically necessary, but do not kill the hairy cells.
People with low neutrophil counts may be given filgrastim or a similar hormone to stimulate production of white blood cells. However, a 1999 study indicates that routine administration of this expensive injected drug has no practical value for HCL patients after cladribine administration.[47] In this study, patients who received filgrastim were just as likely to experience a high fever and to be admitted to the hospital as those who did not, even though the drug artificially inflated their white blood cell counts. This study leaves open the possibility that filgrastim may still be appropriate for patients who have symptoms of infection, or at times other than shortly after cladribine treatment.
Although hairy cells are technically long-lived, instead of rapidly dividing, some late-stage patients are treated with broad-spectrum chemotherapy agents such as methotrexate that are effective at killing rapidly dividing cells. This is not typically attempted unless all other options have been exhausted and it is typically unsuccessful.
Prognosis
Treatment success
More than 95% of new patients are treated well or at least adequately by cladribine or pentostatin.[48] A majority of new patients can expect a disease-free remission time span of about ten years, or sometimes much longer after taking one of these drugs just once. If retreatment is necessary in the future, the drugs are normally effective again, although the average length of remission is somewhat shorter in subsequent treatments. There is also the risk of Shingles, and Peripheral Neuropathy after treatment with cladribine.
As with B-cell chronic lymphocytic leukemia, mutations in the IGHV on hairy cells are associated with better responses to initial treatments and with prolonged survival.[49]
How soon after treatment a patient feels "normal" again depends on several factors, including:
- how advanced the disease was at the time of treatment;
- the patient's underlying health status;
- whether the patient had a "complete response" or only a partial response to the treatment;
- whether the patient experienced any of the rare, but serious side effects such as kidney failure;
- how aggressive the individual's disease is;
- whether the patient is experiencing unusual psychological trauma from the "cancer" diagnosis; and
- how the patient perceived his or her pre-treatment energy level and daily functioning.
Lifespan
With appropriate treatment, the overall projected lifespan for patients is normal or near-normal. In all patients, the first two years after diagnosis have the highest risk for fatal outcome; generally, surviving five years predicts good control of the disease. After five years' clinical remission, patients in the United States with normal blood counts can often qualify for private life insurance with some US companies.[50]
Accurately measuring survival for patients with the variant form of the disease (HCL-V) is complicated by the relatively high median age (70 years old) at diagnosis. However, HCL-V patients routinely survive for more than 10 years, and younger patients can likely expect a long life.
Worldwide, approximately 300 HCL patients per year are expected to die.[51] Some of these patients were diagnosed with HCL due to a serious illness that prevented them from receiving initial treatment in time; many others died after living a normal lifespan and experiencing years of good control of the disease. Perhaps as many as five out of six HCL patients die from some other cause.[original research?]
Follow-up care
Despite decade-long remissions and years of living very normal lives after treatment, hairy cell leukemia is officially considered an incurable disease. While survivors of solid tumors are commonly declared to be permanently cured after two, three, or five years, people who have hairy cell leukemia are never considered 'cured'. Relapses of HCL have happened even after more than twenty years of continuous remission. Patients will require lifelong monitoring and should be aware that the disease can recur even after decades of good health.
While most oncologist consider Hairy Cell Leukemia to be incurable, there is some evidence that some patients are in fact cured after treatments. Of the original 358-patient cohort treated with cladribine at the Scripps Clinic, 9 of 19 in continuous CR for a median of 16 years were free of HCL MRD by flow cytometry and IHC. This suggests that the disease of at least some patients may be cured.[52]
People in remission need regular follow-up examinations after their treatment is over. Most physicians insist on seeing patients at least once a year for the rest of the patient's life, and getting blood counts about twice a year. Regular follow-up care ensures that patients are carefully monitored, any changes in health are discussed, and new or recurrent cancer can be detected and treated as soon as possible. Between regularly scheduled appointments, people who have hairy cell leukemia should report any health problems, especially viral or bacterial infections, as soon as they appear.
HCL patients are also at a slightly higher than average risk for developing a second kind of cancer, such as colon cancer or lung cancer, at some point during their lives (including before their HCL diagnosis). This appears to relate best to the number of hairy cells, and not to different forms of treatment.[53] On average, patients might reasonably expect to have as much as double the risk of developing another cancer, with a peak about two years after HCL diagnosis and falling steadily after that, assuming that the HCL was successfully treated. Aggressive surveillance and prevention efforts are generally warranted, although the lifetime odds of developing a second cancer after HCL diagnosis are still less than 50%.
There is also a higher risk of developing an autoimmune disease.[15] Polyarteritis nodosa has been associated with underlying hairy cell leukemia in certain cases. Autoimmune diseases may also go into remission after treatment of HCL.[15]
Epidemiology
This disease is rare, with fewer than 1 in 10,000 people being diagnosed with HCL during their lives. Men are four to five times more likely to develop hairy cell leukemia than women.[54] In the United States, the annual incidence is approximately 3 cases per 1,000,000 men each year, and 0.6 cases per 1,000,000 women each year.[15]
Most patients are white males over the age of 50,[15] although it has been diagnosed in at least one teenager.[55] It is less common in people of African and Asian descent compared to people of European descent.
It does not appear to be hereditary, although occasional familial cases that suggest a predisposition have been reported,[56] usually showing a common Human Leukocyte Antigen (HLA) type.[15]
Research directions
The Hairy Cell Leukemia Consortium was founded in 2008 to address researchers' concerns about the long-term future of research on the disease.[57] Partly because existing treatments are so successful, the field has attracted very few new researchers.
In 2013 the Hairy Cell Leukemia Foundation Archived 2021-12-22 at the Wayback Machine was created when the Hairy Cell Leukemia Consortium and the Hairy Cell Leukemia Research Foundation joined together. The HCLF is dedicated to improving outcomes for patients by advancing research into the causes and treatment of hairy cell leukemia, as well as by providing educational resources and comfort to all those affected by hairy cell leukemia.[58]
Three immunotoxin drugs have been studied in patients at the NIH National Cancer Institute in the U.S.: BL22,[59] HA22[60] and LMB-2.[61] All of these protein-based drugs combine part of an anti-B cell antibody with a bacterial toxin to kill the cells on internalization. BL22 and HA22 attack a common protein called CD22, which is present on hairy cells and healthy B cells. LMB-2 attacks a protein called CD25, which is not present in HCL-variant, so LMB-2 is only useful for patients with HCL-classic or the Japanese variant. HA-22, now renamed moxetumomab pasudotox, is being studied in patients with relapsed hairy cell leukemia at the National Cancer Institute in Bethesda, Maryland, MD Anderson Cancer Center in Houston, Texas, and Ohio State University in Columbus, Ohio. Other sites for the study are expected to start accepting patients in late 2014, including The Royal Marsden Hospital in London, England.[62]
Other clinical trials[63] are studying the effectiveness of cladribine followed by rituximab in eliminating residual hairy cells that remain after treatment by cladribine or pentostatin. It is not currently known if the elimination of such residual cells will result in more durable remissions.
BRAF mutation has been frequently detected in HCL (Tiacci et al. NEJM 2011) and some patients may respond to Vemurafenib
The major remaining research questions are identifying the cause of HCL and determining what prevents hairy cells from maturing normally.[64]
See also
References
- ↑ "Hairy cell leukemia: MedlinePlus Medical Encyclopedia". medlineplus.gov. Archived from the original on 2018-10-24. Retrieved 2018-04-17.
- ↑ Schrek R, Donnelly WJ (February 1966). ""Hairy" cells in blood in lymphoreticular neoplastic disease and "flagellated" cells of normal lymph nodes". Blood. 27 (2): 199–211. doi:10.1182/blood.V27.2.199.199. PMID 5322749.
- ↑ "Hairy cell leukemia | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 2019-11-15. Retrieved 2018-04-17.
- ↑ "Hairy Cell Leukemia Treatment". National Cancer Institute. Archived from the original on 2008-05-14. Retrieved 2007-09-07.
- ↑ Zuzel M, Cawley JC, Paton RC, Burns GF, McNicol GP (1979). "Platelet function in hairy-cell leukaemia". J. Clin. Pathol. 32 (8): 814–21. doi:10.1136/jcp.32.8.814. PMC 1145815. PMID 512041.
- ↑ Pandolfino J, Hakimian D, Rademaker AW, Tallman MS (July 1997). "Hypocholesterolemia in hairy cell leukemia: a marker for proliferative activity". Am. J. Hematol. 55 (3): 129–33. doi:10.1002/(SICI)1096-8652(199707)55:3<129::AID-AJH2>3.0.CO;2-U. PMID 9256291.
- ↑ Juliusson G, Vitols S, Liliemark J (July 1995). "Mechanisms behind hypocholesterolaemia in hairy cell leukaemia". BMJ. 311 (6996): 27. doi:10.1136/bmj.311.6996.27. PMC 2550084. PMID 7613319.
- ↑ Clavel J, Mandereau L, Cordier S, Le Goaster C, Hémon D, Conso F, Flandrin G (1995). "Hairy cell leukaemia, occupation, and smoking". Br. J. Haematol. 91 (1): 154–61. doi:10.1111/j.1365-2141.1995.tb05261.x. PMID 7577624. S2CID 6423475.
- ↑ Orlandi G, Fanucchi S, Strata G, Pataleo L, Landucci Pellegrini L, Prontera C, Martini A, Murri L (2000). "Transient autonomic nervous system dysfunction during hyperacute stroke". Acta Neurol. Scand. 102 (5): 317–21. doi:10.1034/j.1600-0404.2000.102005317.x. PMID 11083509. S2CID 46685771.
- ↑ Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, Pucciarini A, Bigerna B, Pacini R, Wells VA, Sportoletti P, Pettirossi V, Mannucci R, Elliott O, Liso A, Ambrosetti A, Pulsoni A, Forconi F, Trentin L, Semenzato G, Inghirami G, Capponi M, Di Raimondo F, Patti C, Arcaini L, Musto P, Pileri S, Haferlach C, Schnittger S, Pizzolo G, Foà R, Farinelli L, Haferlach T, Pasqualucci L, Rabadan R, Falini B (2011). "BRAFMutations in Hairy-Cell Leukemia". N Engl J Med. 364 (24): 2305–2315. doi:10.1056/NEJMoa1014209. PMC 3689585. PMID 21663470.
- ↑ Committee to Review the Health Effects in Vietnam Veterans of Exposure to Herbicides (Seventh Biennial Update) (2009). Veterans and Agent Orange: Update 2008. Institute of Medicine. pp. 349–350. doi:10.17226/12662. ISBN 978-0-309-13884-0. PMID 25032343. Archived from the original on 2011-09-28. Retrieved 2021-12-12.
- ↑ Feuer G, Green PL (September 2005). "Comparative biology of human T-cell lymphotropic virus type 1 (HTLV-1) and HTLV-2". Oncogene. 24 (39): 5996–6004. doi:10.1038/sj.onc.1208971. PMC 2659530. PMID 16155606.
- ↑ Rosenblatt JD, Giorgi JV, Golde DW, Ezra JB, Wu A, Winberg CD, Glaspy J, Wachsman W, Chen IS (February 1988). "Integrated human T-cell leukemia virus II genome in CD8 + T cells from a patient with "atypical" hairy cell leukemia: evidence for distinct T and B cell lymphoproliferative disorders". Blood. 71 (2): 363–9. doi:10.1182/blood.V71.2.363.363. PMID 2827811.
{{cite journal}}: CS1 maint: url-status (link) - ↑ Roucoux DF, Murphy EL (2004). "The epidemiology and disease outcomes of human T-lymphotropic virus type II". AIDS Rev. 6 (3): 144–54. PMID 15595431.
- ↑ 15.00 15.01 15.02 15.03 15.04 15.05 15.06 15.07 15.08 15.09 15.10 15.11 15.12 15.13 15.14 15.15 15.16 15.17 15.18 Wintrobe, Maxwell Myer (2004). Wintrobe's clinical hematology. John G. Greer; John Foerster, John N. Lukens, George M Rodgers, Frixos Paraskevas (11 ed.). Hagerstown, MD: Lippincott Williams & Wilkins. pp. 2465–2466. ISBN 978-0-7817-3650-3.
- ↑ 16.0 16.1 Wanko, Sam O.; de Castro, Carlos (July 2006). "Hairy Cell Leukemia: An Elusive but Treatable Disease" (PDF). The Oncologist. 11 (7): 780–789. doi:10.1634/theoncologist.11-7-780. PMID 16880237. Archived (PDF) from the original on 2018-07-20. Retrieved 2021-12-12.
- ↑ Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, Pucciarini A, Bigerna B, Pacini R, Wells VA, Sportoletti P, Pettirossi V, Mannucci R, Elliott O, Liso A, Ambrosetti A, Pulsoni A, Forconi F, Trentin L, Semenzato G, Inghirami G, Capponi M, Di Raimondo F, Patti C, Arcaini L, Musto P, Pileri S, Haferlach C, Schnittger S, Pizzolo G, Foà R, Farinelli L, Haferlach T, Pasqualucci L, Rabadan R, Falini B (June 2011). "BRAF mutations in hairy-cell leukemia". N. Engl. J. Med. 364 (24): 2305–15. doi:10.1056/NEJMoa1014209. PMC 3689585. PMID 21663470.
- ↑ Research on hairy-cell leukaemia shows the promise of new DNA-scanning technologies Archived 2021-03-06 at the Wayback Machine - Cancer Research UK science blog
- ↑ Vanhentenrijk V, De Wolf-Peeters C, Wlodarska I (2004). "Comparative expressed sequence hybridization studies of hairy cell leukemia show uniform expression profile and imprint of spleen signature". Blood. 104 (1): 250–5. doi:10.1182/blood-2004-01-0181. PMID 15016649.
- ↑ "Clinical Flow Cytometry Case #54". Archived from the original on 2007-10-09. Retrieved 2007-09-07.
- ↑ Cannon T, Mobarek D, Wegge J, Tabbara IA (October 2008). "Hairy cell leukemia: current concepts". Cancer Invest. 26 (8): 860–5. doi:10.1080/07357900801965034. PMID 18798068. S2CID 6196898.
- ↑ Hairy Cell Leukemia~Differentials at eMedicine
- ↑ Cessna, Melissa H.; Hartung, Leah; Tripp, Sheryl; Perkins, Sherrie L.; Bahler, David W. (January 2005). "Hairy Cell Leukemia Variant: Fact or Fiction". American Journal of Clinical Pathology. 123 (1): 132–138. doi:10.1309/8QYTYQ1CLQMHQ9CL. PMID 15762289.
- ↑ 24.0 24.1 Sainati L, Matutes E, Mulligan S, de Oliveira MP, Rani S, Lampert IA, Catovsky D (July 1990). "A variant form of hairy cell leukemia resistant to alpha-interferon: clinical and phenotypic characteristics of 17 patients". Blood. 76 (1): 157–62. doi:10.1182/blood.V76.1.157.157. PMID 2364167.
{{cite journal}}: CS1 maint: url-status (link) - ↑ Cawley JC, Burns GF, Hayhoe FG (1980). "A chronic lymphoproliferative disorder with distinctive features: a distinct variant of hairy-cell leukaemia". Leuk. Res. 4 (6): 547–59. doi:10.1016/0145-2126(80)90066-1. PMID 7206776.
- ↑ 26.0 26.1 26.2 26.3 Matutes E, Wotherspoon A, Brito-Babapulle V, Catovsky D (2001). "The natural history and clinico-pathological features of the variant form of hairy cell leukemia". Leukemia. 15 (1): 184–6. doi:10.1038/sj.leu.2401999. PMID 11243388. S2CID 6609635.
- ↑ Ya-In, Charin; Brandwein, Joseph; Pantalony, Dominic; Chang, Hong (1 March 2005). "Hairy Cell Leukemia Variant With Features of Intrasinusoidal Bone Marrow Involvement". Archives of Pathology & Laboratory Medicine. 129 (3): 395–398. doi:10.5858/2005-129-395-HCLVWF. PMID 15737038.
- ↑ Kreitman, Robert J.; Wilson, Wyndham; Calvo, Katherine R.; Arons, Evgeny; Roth, Laura; Sapolsky, Jeffrey; Zhou, Hong; Raffeld, Mark; Stetler-Stevenson, Maryalice (15 December 2013). "Cladribine with Immediate Rituximab for the Treatment of Patients with Variant Hairy Cell Leukemia". Clinical Cancer Research. 19 (24): 6873–6881. doi:10.1158/1078-0432.CCR-13-1752. PMC 3867590. PMID 24277451.
- ↑ "HCL-V". Hairy Cell Leukemia Foundation. Archived from the original on 2016-06-01. Retrieved 2016-05-31.
- ↑ Burthem J, Cawley JC (January 1994). "The bone marrow fibrosis of hairy-cell leukemia is caused by the synthesis and assembly of a fibronectin matrix by the hairy cells". Blood. 83 (2): 497–504. doi:10.1182/blood.V83.2.497.497. PMID 8286747.
{{cite journal}}: CS1 maint: url-status (link) - ↑ de Totero D, Tazzari PL, Lauria F, Raspadori D, di Celle PF, Carbone A, Gobbi M, Foa R (July 1993). "Phenotypic analysis of hairy cell leukemia: "variant" cases express the interleukin-2 receptor beta chain, but not the alpha chain (CD25)". Blood. 82 (2): 528–35. doi:10.1182/blood.V82.2.528.528. PMID 8329708.
{{cite journal}}: CS1 maint: url-status (link) - ↑ Matutes E, Wotherspoon A, Catovsky D (2003). "The variant form of hairy-cell leukaemia". Best Pract Res Clin Haematol. 16 (1): 41–56. doi:10.1016/S1521-6926(02)00086-5. PMID 12670464.
- ↑ Vallianatou K, Brito-Babapulle V, Matutes E, Atkinson S, Catovsky D (1999). "p53 gene deletion and trisomy 12 in hairy cell leukemia and its variant". Leuk. Res. 23 (11): 1041–5. doi:10.1016/S0145-2126(99)00127-7. PMID 10576509.
- ↑ Arons, Evgeny; Suntum, Tara; Stetler-Stevenson, Maryalice; Kreitman, Robert J. (19 November 2009). "VH4-34+ hairy cell leukemia, a new variant with poor prognosis despite standard therapy". Blood. 114 (21): 4687–4695. doi:10.1182/blood-2009-01-201731. PMC 2780305. PMID 19745070.
- ↑ 宮崎, 睦子; 田口, 昭彦; 櫻木, 志津; 三谷, 紀之; 松田, 万幸; 篠原, 健次 (2004). "Cladribine投与が奏効したhairy cell leukeimia, Japanese variant" [Hairy cell leukemia, Japanese variant, successfully treated with cladribine]. 臨床血液 (in 日本語). 45 (5): 405–407. doi:10.11406/rinketsu.45.405. PMID 15199752.
- ↑ Ravandi F, Jorgensen JL, O'Brien SM, Verstovsek S, Koller CA, Faderl S, Giles FJ, Ferrajoli A, Wierda WG, Odinga S, Huang X, Thomas DA, Freireich EJ, Jones D, Keating MJ, Kantarjian HM (June 2006). "Eradication of minimal residual disease in hairy cell leukemia". Blood. 107 (12): 4658–62. doi:10.1182/blood-2005-11-4590. PMID 16497968.
- ↑ Robak T, Jamroziak K, Gora-Tybor J, Blonski JZ, Kasznicki M, Dwilewicz-Trojaczek J, Wiater E, Zdunczyk A, Dybowicz J, Dmoszynska A, Wojtaszko M, Zdziarska B, Calbecka M, Kostyra A, Hellmann A, Lewandowski K, Stella-Holowiecka B, Sulek K, Gawronski K, Skotnicki AB, Nowak W, Zawilska K, Molendowicz-Portala L, Kloczko J, Sokolowski J, Warzocha K, Seferynska I, Ceglarek B, Konopka L (2007). "Cladribine in a weekly versus daily schedule for untreated active hairy cell leukemia: final report from the Polish Adult Leukemia Group (PALG) of a prospective, randomized, multicenter trial". Blood. 109 (9): 3672–5. doi:10.1182/blood-2006-08-042929. PMID 17209059. Archived from the original on 2007-09-29. Retrieved 2007-07-04.
- ↑ Else M, Ruchlemer R, Osuji N, Del Giudice I, Matutes E, Woodman A, Wotherspoon A, Swansbury J, Dearden C, Catovsky D (2005). "Long remissions in hairy cell leukemia with purine analogs: a report of 219 patients with a median follow-up of 12.5 years". Cancer. 104 (11): 2442–8. doi:10.1002/cncr.21447. PMID 16245328. S2CID 43282431.
- ↑ Saven, Alan; Piro, LD (15 August 1993). "Complete Remissions in Hairy Cell Leukemia with 2-Chlorodeoxyadenosine after Failure with 2-Deoxycoformycin". Annals of Internal Medicine. 119 (4): 278–283. doi:10.7326/0003-4819-119-4-199308150-00005. PMID 8101069. S2CID 34812778.
- ↑ Thomas, Deborah A.; O'Brien, Susan; Bueso-Ramos, Carlos; Faderl, Stefan; Keating, Michael J.; Giles, Francis J.; Cortes, Jorge; Kantarjian, Hagop M. (1 December 2003). "Rituximab in relapsed or refractory hairy cell leukemia". Blood. 102 (12): 3906–3911. doi:10.1182/blood-2003-02-0630. PMID 12816862.
- ↑ Nieva, Jorge; Bethel, Kelly; Saven, Alan (1 August 2003). "Phase 2 study of rituximab in the treatment of cladribine-failed patients with hairy cell leukemia". Blood. 102 (3): 810–813. doi:10.1182/blood-2003-01-0014. PMID 12663446.
- ↑ Narat S, Gandla J, Dogan A, Mehta A (August 2005). "Successful treatment of hairy cell leukemia variant with rituximab". Leuk. Lymphoma. 46 (8): 1229–32. doi:10.1080/10428190500083433. PMID 16085567. S2CID 20116705.
- ↑ Hairy Cell Leukemia~medication at eMedicine
- ↑ Quesada JR, Reuben J, Manning JT, Hersh EM, Gutterman JU (January 1984). "Alpha interferon for induction of remission in hairy-cell leukemia". N. Engl. J. Med. 310 (1): 15–8. doi:10.1056/NEJM198401053100104. PMID 6689734.
- ↑ 45.0 45.1 Ratain MJ, Golomb HM, Vardiman JW, Vokes EE, Jacobs RH, Daly K (1985). "Treatment of hairy cell leukemia with recombinant alpha 2 interferon". Blood. 65 (3): 644–8. doi:10.1182/blood.V65.3.644.644. PMID 3971043.
- ↑ 46.0 46.1 Baker PK, Pettitt AR, Slupsky JR, Chen HJ, Glenn MA, Zuzel M, Cawley JC (2002). "Response of hairy cells to IFN-alpha involves induction of apoptosis through autocrine TNF-alpha and protection by adhesion". Blood. 100 (2): 647–53. doi:10.1182/blood.V100.2.647. PMID 12091360. S2CID 6712599. Archived from the original on 2022-02-13. Retrieved 2021-12-12.
- ↑ Saven, Alan; Burian, Carol; Adusumalli, Jasvant; Koziol, James A. (15 April 1999). "Filgrastim for Cladribine-Induced Neutropenic Fever in Patients With Hairy Cell Leukemia". Blood. 93 (8): 2471–2477. doi:10.1182/blood.V93.8.2471. PMID 10194424.
- ↑ Else M, Ruchlemer R, Osuji N, Del Giudice I, Matutes E, Woodman A, Wotherspoon A, Swansbury J, Dearden C, Catovsky D (December 2005). "Long remissions in hairy cell leukemia with purine analogs: a report of 219 patients with a median follow-up of 12.5 years". Cancer. 104 (11): 2442–8. doi:10.1002/cncr.21447. PMID 16245328. S2CID 43282431.
- ↑ Forconi F, Sozzi E, Cencini E, Zaja F, Intermesoli T, Stelitano C, Rigacci L, Gherlinzoni F, Cantaffa R, Baraldi A, Gallamini A, Zaccaria A, Pulsoni A, Gobbi M, Tassi M, Raspadori D, Leoncini L, Rinaldi A, Sabattini E, Bertoni F, Pileri SA, Lauria F (November 2009). "Hairy cell leukemias with unmutated IGHV genes define the minor subset refractory to single agent cladribine and with more aggressive behavior". Blood. 114 (21): 4696–4702. doi:10.1182/blood-2009-03-212449. PMID 19667403. Archived from the original on 2022-02-13. Retrieved 2021-12-12.
- ↑ Brown, Richard T. (August 2002). "Hairy Cell Leukemia" (PDF). Brokerage Bulletin. 7 (5): 3. Archived from the original (PDF) on 2012-02-24. Retrieved 2018-11-29.[unreliable medical source?]
- ↑ "Archived copy". Archived from the original on 2007-08-13. Retrieved 2007-07-30.
{{cite web}}: CS1 maint: archived copy as title (link) - ↑ "Update on Hairy Cell Leukemia – Hematology & Oncology". Archived from the original on 2020-07-19. Retrieved 2021-12-12.
- ↑ Au WY, Klasa RJ, Gallagher R, Le N, Gascoyne RD, Connors JM (August 1998). "Second malignancies in patients with hairy cell leukemia in British columbia: a 20-year experience". Blood. 92 (4): 1160–4. doi:10.1182/blood.V92.4.1160. PMID 9694703.
{{cite journal}}: CS1 maint: url-status (link) - ↑ "Hairy cell leukemia". Archived from the original on 2007-09-14. Retrieved 2007-09-07.
- ↑ Yetgin S, Olcay L, Yenicesu I, Oner AF, Cağlar M (September 2001). "Relapse in hairy cell leukemia due to isolated nodular skin infiltration". Pediatr Hematol Oncol. 18 (6): 415–7. doi:10.1080/088800101316922047. PMID 11554237. S2CID 793191.
- ↑ Gramatovici M, Bennett JM, Hiscock JG, Grewal KS (April 1993). "Three cases of familial hairy cell leukemia". Am. J. Hematol. 42 (4): 337–9. doi:10.1002/ajh.2830420402. PMID 8493982. S2CID 34449485.
- ↑ Holzman D (March 2009). "Has success spoiled hairy cell leukemia research? Key questions go unanswered, despite big gains". J. Natl. Cancer Inst. 101 (6): 370–3. doi:10.1093/jnci/djp051. PMID 19276454.
- ↑ "| Hairy Cell Leukemia Foundation". Hairycellleukemia.org. 2014-06-20. Archived from the original on 2021-12-22. Retrieved 2015-12-24.
- ↑ Clinical trial number NCT00074048 for "BL22 Immunotoxin in Treating Patients Previously Treated With Cladribine for Hairy Cell Leukemia" at ClinicalTrials.gov
- ↑ Clinical trial number NCT00462189 for "Safety Study of CAT-8015 Immunooxin in Patients With HCL With Advance Disease" at ClinicalTrials.gov
- ↑ Clinical trial number NCT00337311 for "LMB-2 to Treat Hairy Cell Leukemia" at ClinicalTrials.gov
- ↑ "Moxetumomab Pasudotox for Advanced Hairy Cell Leukemia - Full Text View". ClinicalTrials.gov. Archived from the original on 2015-12-24. Retrieved 2015-12-24.
- ↑ For example, trial NCT00412594 Archived 2011-06-13 at the Wayback Machine and NCT00923013 Archived 2012-04-05 at the Wayback Machine
- ↑ Cawley, John. "The Biology of the Disease Including Unanswered Questions | HairyCell.org". Archived from the original on 2011-07-26. Retrieved 2010-06-23.
External links
| Classification | |
|---|---|
| External resources |
- Pages with script errors
- CS1 maint: url-status
- Webarchive template wayback links
- CS1 日本語-language sources (ja)
- All articles lacking reliable references
- Articles lacking reliable references from December 2021
- Articles with invalid date parameter in template
- CS1 maint: archived copy as title
- All articles with unsourced statements
- Articles with unsourced statements from November 2021
- Articles with unsourced statements from December 2021
- All articles that may contain original research
- Articles that may contain original research from September 2015
- Lymphocytic leukemia
- Lymphoid-related cutaneous conditions