Angioimmunoblastic T-cell lymphoma
| Angioimmunoblastic T-cell lymphoma | |
|---|---|
| Other names: immunoblastic lymphadenopathy (Lukes-Collins Classification), AILD-type (lymphogranulomatosis X) T-cell lymphoma (Kiel Classification)[1] | |
| Specialty | Hematology and oncology |
Angioimmunoblastic T-cell lymphoma (AITL, sometimes misspelled AILT) (formerly known as "angioimmunoblastic lymphadenopathy with dysproteinemia"[2]: 747 ) is a mature T-cell lymphoma of blood or lymph vessel immunoblasts characterized by a polymorphous lymph node infiltrate showing a marked increase in follicular dendritic cells (FDCs) and high endothelial venules (HEVs) and systemic involvement.[1]
Signs and symptoms
Patients with this disease usually present at an advanced stage and show systemic involvement. The clinical findings typically include a pruritic skin rash and possibly edema, ascites, pleural effusions, and arthritis.[3][4]
Sites of involvement
Due to the systemic nature of this disease, neoplastic cells can be found in lymph nodes, liver, spleen, skin, and bone marrow.[citation needed]
Causes
This disease was originally thought to be a premalignant condition, termed angioimmunoblastic lymphadenopathy, and this atypical reactive lymphadenopathy carried a risk for transformation into a lymphoma. Currently, it is postulated that the originating cell for this disease is a mature (post-thymic) CD4+ T-cell that arises de novo,[1] although some researchers argue that there is a premalignant subtype of this disease.[5][6] The Epstein–Barr virus (EBV) is observed in the majority of cases,[1] being identified in the reactive (i.e. non-malignant) B-cells that comprise part of the polymorphous infiltrate of this disease.[7] These EBV+ B cells have numerous non-malignant crippling mutations, often proliferate excessively, and in some cases may transform into EBV+ B cell lymphomas. The other cell types in these infiltrates, including the malignant TFH cells, are EBV negative. While the World Health Organization (2016) has classified these EBV-associated cases as one of the Epstein-Barr virus-associated lymphoproliferative diseases (see EBV+ angioimmunoblastic T cell lymphoma, the role of the virus in the development and/or progression of EBV+ angioimmunoblastic T cell lymphoma is unclear.[8] Immunodeficiency is also seen with this disease, but it is a sequela to the condition and not a predisposing factor.[1]
Diagnosis
-
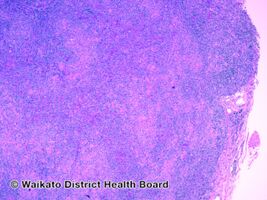
Angioimmunoblastic T-cell lymphoma-pathology
-
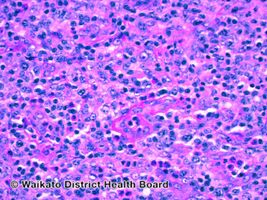
Angioimmunoblastic T-cell lymphoma-pathology
-

Angioimmunoblastic T-cell lymphoma-pathology
.jpg)
.jpg)
.jpg)
Laboratory findings
The classical laboratory finding is polyclonal hypergammaglobulinemia, and other immunoglobulin derangements are also seen, including hemolytic anemia with cold agglutinins, circulating immune complexes, anti-smooth muscle antibodies, and positive rheumatoid factor.[1][3]
Lymph node
The normal architecture of a lymph node is partially effaced by a polymorphous infiltrate and residual follicles are commonly seen. The polymorphous infiltrate consists of lymphocytes of moderate size with pale/clear cytoplasm and smaller reactive lymphocytes, eosinophils, histiocytes, plasma cells, and follicular dendritic cells. In addition, blast-like B-cells are occasionally seen. A classic morphological finding is the aborization and proliferation of high endothelial venules.[1] Hyperplastic germinal centers and Reed-Sternberg-like cells can also be seen.[9][10]
Immunophenotype
AITL typically has the phenotype of a mixture of CD4+ and CD8+ T-cells, with a CD4:CD8 ratio greater than unity. Polyclonal plasma cells and CD21+ follicular dendritic cells are also seen.[1]
Molecular findings
Clonal T-cell receptor gene rearrangements are detected in 75% of cases,[11] and immunoglobin gene rearrangements are seen in 10% of cases, and these cases are believed to be due to expanded EBV-driven B-cell populations.[12] Similarly, EBV-related sequences can be detected in most cases, usually in B-cells but occasionally in T-cells.[7][13] Trisomy 3, trisomy 5, and +X are the most frequent chromosomal abnormalities found in AITL cases.[14][15]
Treatment
There is no proven or standard first-line chemotherapy that works for the majority of AITL patients. There are several clinical trials that offer treatment options that can fight the disease. Stem cell transplantation is the treatment of choice, with the allogeneic one being the preference because AITL tends to recur after autologous transplants.[citation needed]
Epidemiology
The typical patient with angioimmunoblastic T-cell lymphoma (AITL) is either middle-aged or elderly, and no gender preference for this disease has been observed.[1] AITL comprises 15–20% of peripheral T-cell lymphomas and 1–2% of all non-Hodgkin lymphomas.[16]
See also
References
- ↑ 1.0 1.1 1.2 1.3 1.4 1.5 1.6 1.7 1.8 Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W (2008). "11 Mature T- and NK-cell neoplasms: Angioimmunoblastic T-cell lymphoma". WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC WHO Classification of Tumours. Vol. 2 (4th ed.). IARC. ISBN 978-9283224310. Archived from the original on 2017-06-05. Retrieved 2021-03-29.
- ↑ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: Clinical Dermatology. Saunders Elsevier. ISBN 0-7216-2921-0.
- ↑ 3.0 3.1 Siegert W, Nerl C, Agthe A, et al. (September 1995). "Angioimmunoblastic lymphadenopathy (AILD)-type T-cell lymphoma: prognostic impact of clinical observations and laboratory findings at presentation. The Kiel Lymphoma Study Group". Ann. Oncol. 6 (7): 659–64. doi:10.1093/oxfordjournals.annonc.a059281. PMID 8664186.
- ↑ Jaffe ES (September 1995). "Angioimmunoblastic T-cell lymphoma: new insights, but the clinical challenge remains". Ann. Oncol. 6 (7): 631–2. doi:10.1093/oxfordjournals.annonc.a059273. PMID 8664181.
- ↑ Frizzera G, Kaneko Y, Sakurai M (January 1989). "Angioimmunoblastic lymphadenopathy and related disorders: a retrospective look in search of definitions". Leukemia. 3 (1): 1–5. PMID 2642571.
- ↑ Smith JL, Hodges E, Quin CT, McCarthy KP, Wright DH (February 2000). "Frequent T and B Cell Oligoclones in Histologically and Immunophenotypically Characterized Angioimmunoblastic Lymphadenopathy". Am. J. Pathol. 156 (2): 661–9. doi:10.1016/S0002-9440(10)64770-0. PMC 1850038. PMID 10666395.
- ↑ 7.0 7.1 Weiss LM, Jaffe ES, Liu XF, Chen YY, Shibata D, Medeiros LJ (April 1992). "Detection and localization of Epstein-Barr viral genomes in angioimmunoblastic lymphadenopathy and angioimmunoblastic lymphadenopathy-like lymphoma". Blood. 79 (7): 1789–95. doi:10.1182/blood.V79.7.1789.1789. PMID 1373088. Archived from the original on 2021-08-27. Retrieved 2021-03-29.
- ↑ Rezk SA, Zhao X, Weiss LM (September 2018). "Epstein-Barr virus (EBV)-associated lymphoid proliferations, a 2018 update". Human Pathology. 79: 18–41. doi:10.1016/j.humpath.2018.05.020. PMID 29885408.
- ↑ Quintanilla-Martinez L, Fend F, Moguel LR, et al. (October 1999). "Peripheral T-cell lymphoma with Reed–Sternberg-like cells of B-cell phenotype and genotype associated with Epstein–Barr virus infection". Am. J. Surg. Pathol. 23 (10): 1233–40. doi:10.1097/00000478-199910000-00008. PMID 10524524.
- ↑ Ree HJ, Kadin ME, Kikuchi M, et al. (June 1998). "Angioimmunoblastic lymphoma (AILD-type T-cell lymphoma) with hyperplastic germinal centers". Am. J. Surg. Pathol. 22 (6): 643–55. doi:10.1097/00000478-199806000-00001. PMID 9630171.
- ↑ Feller AC, Griesser H, Schilling CV, et al. (December 1988). "Clonal gene rearrangement patterns correlate with immunophenotype and clinical parameters in patients with angioimmunoblastic lymphadenopathy". Am. J. Pathol. 133 (3): 549–56. PMC 1880823. PMID 2849301.
- ↑ Lipford EH, Smith HR, Pittaluga S, Jaffe ES, Steinberg AD, Cossman J (February 1987). "Clonality of angioimmunoblastic lymphadenopathy and implications for its evolution to malignant lymphoma". J. Clin. Invest. 79 (2): 637–42. doi:10.1172/JCI112860. PMC 424152. PMID 3805286.
- ↑ Anagnostopoulos I, Hummel M, Finn T, et al. (October 1992). "Heterogeneous Epstein-Barr virus infection patterns in peripheral T-cell lymphoma of angioimmunoblastic lymphadenopathy type". Blood. 80 (7): 1804–12. doi:10.1182/blood.V80.7.1804.1804. PMID 1327284. Archived from the original on 2021-08-27. Retrieved 2021-03-29.
- ↑ Kaneko Y, Maseki N, Sakurai M, et al. (August 1988). "Characteristic karyotypic pattern in T-cell lymphoproliferative disorders with reactive "angioimmunoblastic lymphadenopathy with dysproteinemia-type" features". Blood. 72 (2): 413–21. doi:10.1182/blood.V72.2.413.413. PMID 3261178. Archived from the original on 2021-08-27. Retrieved 2021-03-29.
- ↑ Schlegelberger B, Zhang Y, Weber-Matthiesen K, Grote W (October 1994). "Detection of aberrant clones in nearly all cases of angioimmunoblastic lymphadenopathy with dysproteinemia-type T-cell lymphoma by combined interphase and metaphase cytogenetics". Blood. 84 (8): 2640–8. doi:10.1182/blood.V84.8.2640.2640. PMID 7919378. Archived from the original on 2021-08-27. Retrieved 2021-03-29.
- ↑ "A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin's lymphoma. The Non-Hodgkin's Lymphoma Classification Project". Blood. 89 (11): 3909–18. June 1997. doi:10.1182/blood.V89.11.3909. PMID 9166827. Archived from the original on 2021-08-27. Retrieved 2021-03-29.
External links
| Classification | |
|---|---|
| External resources |
- Pages with script errors
- Articles with hatnote templates targeting a nonexistent page
- All articles with unsourced statements
- Articles with unsourced statements from March 2021
- Articles with invalid date parameter in template
- Lymphoid-related cutaneous conditions
- Lymphoma
- Epstein–Barr virus-associated diseases