Waardenburg Syndrome Type 1
| Waardenburg Syndrome Type 1 | |
|---|---|
 | |
| White forelock hair is shown here. When the Waardenburg Syndrome Type 1 first was studied, this feature along with the dystopia canthorum was the main focus of the research and identifying factor for patients. | |
| Causes | PAX3 gene mutation |
| Frequency | Lua error in Module:PrevalenceData at line 5: attempt to index field 'wikibase' (a nil value). |
Waardenburg Syndrome Type 1 is a congenital disorder that caused by a mutation in the PAX3 gene that results in abnormal development in the neural crest during early development. Type 1 results in early graying and white forelock and a notable distance between the eyes, noted as dystopia canthorum. Common symptoms of the disease also includes non-progressive hearing loss in majority of patients with Type 1. Patients can display complete or partial heterochromia and hypoplastic blue irides and congenital leukemia.[1]
Signs and symptoms
Type 1 of the Waardenburg Syndrome’s notable feature is dystopia canthorum. Along with this feature, some patients' eyelids are fused medially, resulting in medial sclerae. Inferior lachrymal is moved laterally, along with punctae opposite of the cornea.[2] Other features include high and broad nasal root and also nasi hypoplasia. A squared jaw is reported in some patients. Others present with spina bifida due to the mutation in the neural crest during early development. Other features include the white forelock and graying that occurs in most patients prior to reaching age thirty. In extremely rare cases, the forelock may even be colored red rather than white. The coloring of the forelock may differ from patient to patient. It can appear at birth, or later in the life of those affected and size may vary. It is usually observed in the midline of the hair, but it can appear in other locations as well.[1] Associated with the hair forelock, skin pigmentation can also appear in different parts of the body and limbs (suspected to be caused by the mutation in KIT). In parts of the body of some patients, congenital leukoderma is observed.[1]
Similar to other types of the syndrome, Type 1 displays heterochromia in some patients, sometimes complete or partial. If it happens to be partial, the differently colored iris is separated from the radial segment of the eye. Deficient iris stroma along with hypoplastic blue arises have also been found in some patients with Type 1.[citation needed] Another one of the symptoms is hearing loss, presenting in about 69% of cases.[2] The types of hearing loss reported in patients differ over a great spectrum. Some of which could be bilateral or unilateral, sensorineural sometimes a combination of different ones in each ear. The most common loss types reported for Type 1 are profound and bilateral. Waardenburg Type 1 is estimated to cause 3% of overall congenital deaf children.[1]
-
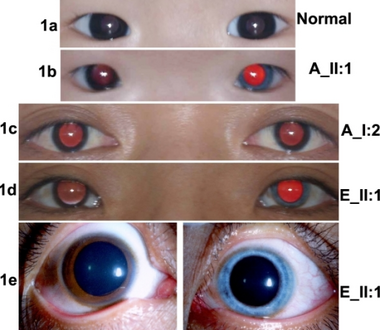
a-e)Images of eyes from WS1 individuals and controls
-

A person displaying dystopia canthorum as well as the hypoplastic blue arises, which are both physical features associated with Type 1 Waardenburg Syndrome.


Genetics
Type 1 Waardenburg Syndrome is a dominant autosomal disease, with extremely high penetrance of nearly 85% [1] Type 1, as well as type 3, are linked to a mutation in the PAX3 gene located in chromosome 2q35. This gene is responsible for encoding a transaction factor named Drosophila paired (noted as prd) resulting in essential proteins. These proteins are responsible for multiple functions in early human development such as stem cell pluripotency, as well as inhibition of differentiation when needed and cell-lineage specification, migration, and proliferation in the cells . These functions are essential in properly developing the central nervous system, somites, skeletal muscles, and neural crest-derived cells that are present throughout various cell types in the body. PAX3 controls the neural crest development through regulation of c-RET, TGF-b2, and WNT1 which are essential controllers of migration and differentiation.[3]
PAX3 in combination with other transcription factors such as MITF and TRP1 can control melanocyte development. This gene also plays a major role in muscle development as it regulated myoD and myf-5, essential transcription factors in muscle development.[4]
The structure of the gene and protein are well understood in research, PAX3 consists of 10 exons and results in major proteins that has 479 amino acids. It has also been found that a majority of the mutations that occur on the PAX3 gene is located on the 2-6 exons. Heterozygous mutation is the basis of the majority of Type 1 Waardenburg Syndrome mutations.[3]
Diagnosis
Type 1 Waardenburg Syndrome is sometimes misdiagnosed as Type 2, therefore when measuring dystopia canthorum if present in a certain patient has been set in strict guidelines. There is a biometric index used as well formula to create a discriminant analysis. These measurements are based on three indices, based on the distances of the inner canthal, interpupillary, and outer canthal.[5] This testing of the dystopia canthorum is referred to as the W index. Other features and dysfunctions are used for diagnoses such as hearing loss or pigmentation of the skin and the notable forelock that appears in the hair. A diagnosis can also be provided through genetic testing and identification of the PAX3 mutation in the patient’s genes. Gene analysis can take on a variety of tests such as single-gene testing (focus on PAX3), a multigene panel (PAX3 along with other genes), and a more comprehensive genomic testing when available.[6]
A list of major criteria was created to assess patients for Type 1 Waardenburg Syndrome. The list includes similar features as listed above such as congenital sensorineural hearing loss, white forelock, and hair pigmentation, pigmentation abnormality of the iris, dystopia canthorum, and related family members who exhibit the disease.[7]
Management
There is no direct treatment for the patients with Waardenburg Syndrome Type 1, however, there are multiple ways in which the symptoms are managed. There are some options for hearing loss aid depending on the type faced by the patient. In previous cases, cochlea implants were successful to aid the hearing loss.[1] There is also some genetic screening available that can assess whether children can inherit the mutation in the PAX3 gene, but not an overall prediction on the manifestation of the disease in the future generations.[1] Pregnant women who are at risk for children suffering from this disease are recommended Folic acid supplementation to assist with the neural crest development.[1] Further counseling is available with life decisions such as pregnancies and starting families. Since Type 1 is a dominant autosomal disease, any offsprings of those affected are likely to also have the disease.
History
Waardenburg syndrome was first described by Petrus J Waardenburg in 1951 in the American Journal of Human Genetics. It is now commonly thought that the description refers to Waardenburg syndrome Type 1. The description focused on the distinguishing feature of dystopia canthorum, the wide distance between the eyes observed in over 80% of patients with Type 1.[1][2] Variations in Type 1 and the difference from Type 2 were later described by Arias in 1971. In 1991, Da-Silva in focused on Brazilian children who exhibited the physical characteristics of Type 1 followed by Winship and Beighton in 1992 with 68 children to describe the onset and manifestation of Type 1.[8] Research in 2016 resulted in connecting the hearing loss and other physical features to the syndrome and provided more accurate diagnosis.[9]
References
- ↑ 1.0 1.1 1.2 1.3 1.4 1.5 1.6 1.7 1.8 Milunsky, Jeff Mark (1993), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "Waardenburg Syndrome Type I", GeneReviews®, University of Washington, Seattle, PMID 20301703, archived from the original on 2021-02-13, retrieved 2020-05-01
- ↑ 2.0 2.1 2.2 Newton, V E (August 1, 1997). "Waardenburg syndrome". Journal of Medical Genetics. 34 (8): 565–575. doi:10.1136/jmg.34.8.656. PMC 1051028. PMID 9279758 – via PubMed.
- ↑ 3.0 3.1 Pingault, Véronique; Ente, Dorothée; Dastot-Le Moal, Florence; Goossens, Michel; Marlin, Sandrine; Bondurand, Nadège (April 2010). "Review and update of mutations causing Waardenburg syndrome". Human Mutation. 31 (4): 391–406. doi:10.1002/humu.21211. ISSN 1098-1004. PMID 20127975.
- ↑ Watanabe, Atsushi; Takeda, Kazuhisa; Ploplis, Barbara; Tachibana, Masayoshi (March 1998). "Epistatic relationship between Waardenburg Syndrome genes MITF and PAX3". Nature Genetics. 18 (3): 283–286. doi:10.1038/ng0398-283. ISSN 1546-1718. PMID 9500554. S2CID 19631822. Archived from the original on 2022-01-20. Retrieved 2022-02-05.
- ↑ Laestadius, Nancy D.; Aase, Jon M.; Smith, David W. (March 1969). "Normal inner canthal and outer orbital dimensions". The Journal of Pediatrics. 74 (3): 465–468. doi:10.1016/s0022-3476(69)80206-4. ISSN 0022-3476. PMID 5764779.
- ↑ Pardono, Eliete; Bever, Yolande van; Ende, Jenneke van den; Havrenne, Poti C.; Iughetti, Paula; Maestrelli, Sylvia R. P.; F, Orozimbo Costa; Richieri‐Costa, Antonio; Frota‐Pessoa, Oswaldo; Otto, Paulo A. (2003). "Waardenburg syndrome: Clinical differentiation between types I and II". American Journal of Medical Genetics Part A. 117A (3): 223–235. doi:10.1002/ajmg.a.10193. ISSN 1552-4833. PMID 12599185. S2CID 34338079.
- ↑ Kapur, Saroj; Karam, Susan (1991). "Germ-line mosaicism in Waardenburg syndrome". Clinical Genetics. 39 (3): 194–198. doi:10.1111/j.1399-0004.1991.tb03011.x. ISSN 1399-0004. PMID 2036740. S2CID 28888391.
- ↑ Winsbip, Ingrid; Brighton, Peter (1992). "Phenotypic discriminants in the Waardenburg syndrome". Clinical Genetics. 41 (4): 181–188. doi:10.1111/j.1399-0004.1992.tb03660.x. ISSN 1399-0004. PMID 1576755.
- ↑ Da‐Silva, Elias O. (1991). "Waardenburg I syndrome: A clinical and genetic study of two large Brazilian kindreds, and literature review". American Journal of Medical Genetics. 40 (1): 65–74. doi:10.1002/ajmg.1320400113. ISSN 1096-8628. PMID 1887852.