Mixed connective tissue disease
| Mixed connective tissue disease | |
|---|---|
| Other names: Sharp's syndrome[1] | |
| Differential diagnosis | CPT2. |
Mixed connective tissue disease commonly abbreviated as MCTD, is an autoimmune disease characterized by the presence of elevated blood levels of a specific autoantibody, now called anti-U1 ribonucleoprotein (RNP) together with a mix of symptoms of systemic lupus erythematosus (SLE), scleroderma, and polymyositis.[2] The idea behind the "mixed" disease is that this specific autoantibody is also present in other autoimmune diseases such as systemic lupus erythematosus, polymyositis, scleroderma, etc. MCTD was characterized as an individual disease in 1972 by Sharp et al.,[3][4] and the term was introduced by Leroy[5] in 1980.[6]
It is sometimes said to be the same as undifferentiated connective tissue disease,[1] but other experts specifically reject this idea[7] because undifferentiated connective tissue disease is not necessarily associated with serum antibodies directed against the U1-RNP, and MCTD is associated with a more clearly defined set of signs/symptoms.[7]
Signs and symptoms
MCTD combines features of scleroderma, myositis, systemic lupus erythematosus, and rheumatoid arthritis[8] (with some sources adding polymyositis, dermatomyositis, and inclusion body myositis)[9] and is thus considered an overlap syndrome.
The initial clinical manifestations of MCTD usually are unspecific, they can consist of general malaise, arthralgias, myalgias, and fever. The specific signs to suspect this disease is the presence of positive antinuclear antibodies (ANA), specifically anti-RNP, associated with Raynaud's phenomenon.[4] Almost every organ can be affected by MCTD.[2] Raynaud's phenomenon is the most common presenting symptom seen in patients, with arthralgia and swollen hands being the second and third most common respectively.[3] With patients that meet full criteria for MCTD, arthritis is the most common symptom with Raynaud's, swollen hands, leukopenia/lymphopenia, and heartburn following in descending order. A 2016 epidemiological population based study found 3.6 years to be the average amount of time from the first manifestations of the disease until all the criteria for diagnosis were met.[3]
Manifestations include:
- Skin: Raynaud's phenomenon is universal and almost always is present at the beginning of the disease course. The absence puts the diagnosis in question. The capillary alterations are similar to that of scleroderma. Other cutaneous alterations can be observed similar to the types observed in LES and scleroderma.[citation needed]
- Arthritis: Swollen fingers and occasionally diffuse edema are distinctive signs. Arthritis usually is more frequent and severe than that observed in SLE. Approximately 60% present with an obvious arthritis, with deformities similar to those observed in rheumatoid arthritis.[citation needed]
- Myositis: Myalgias – muscular aches and pains – are common, but in the majority of patients muscular debilitation, electromyographic alterations, and elevations in muscular enzymes, like in pure polymyositis are not observed.[citation needed]
- Cardiac disease: Pericarditis is the most common cardiac manifestation, observed in 10–30% of patients. Myocardial involvement can also be observed, usually secondary to pulmonary hypertension, as well as conduction anomalies.
- Pulmonary involvement: Is observed in 75% of patients. It can present as pleural effusion, pulmonary hypertension, interstitial lung disease, thromboembolic disease, and others.[citation needed]
- Renal disease: The absence of severe renal disease is a marker of MCTD. Membranous nephropathy can be observed in some cases.[citation needed]
- Gastrointestinal disease: The most common change is the alteration of esophageal motility like that observed in scleroderma.
- Central nervous system involvement (CNS): The original description of this disease stressed the absence of changes to the CNS, however, there have been trigeminal neuropathies (cranial nerve V), sensorineural hearing loss, and headaches observed in patients with MCTD. [citation needed]
- Hematologic anomalies: The presence of mild anemia and hypergammaglobulinemia are common, other hematologic anomalies such as those observed in SLE can also be observed.[citation needed]
- Laboratory value changes: Rheumatoid factor is positive in 50–70% of patients, and anti–citrullinated protein antibody is detected in 50% of patients. The universal serological findings in patients with MCTD is the presence of anti-nuclear antibody, with anti-nRNP specificity, especially antibodies against protein 68 kD.[citation needed]
-
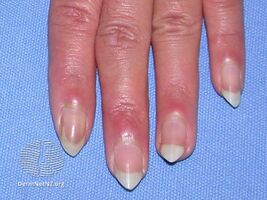
Mixed connective tissue disease
-
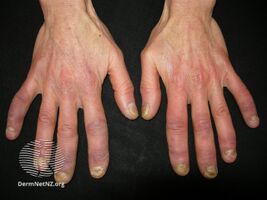
Mixed connective tissue disease
-
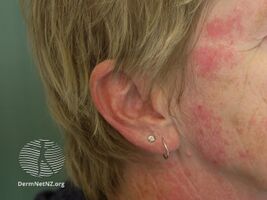
Mixed connective tissue disease
.jpg)
.jpg)
.jpg)
Genetics
The contribution of genetics toward developing MCTD is unknown.[10] Family members have been known to develop MCTD suggesting that genetics may play a role in MCTD, however most cases present individually.[11] As MCTD can present with comorbid connective tissue diseases there must be a genetic link, however it has not yet been discovered. DNA methylation may affect the as yet unknown genetic risks of this disease as patients with MCTD have decreased DNA methylation levels in opposition to their healthy counterparts.[citation needed]
Pathophysiology
MCTD is an autoimmune disorder. Anti-RNP antibodies develop against RNP when RNP is found outside of the nucleus. RNP is immunologically protected due to its location, however if a cell dies and RNP is no longer contained in the nucleus and thus unprotected, the immune system can respond by forming antibodies due to cellular mimicry. Risk to develop MCTD can increase if the body has exposed to molecules or viruses with a similar structure to RNP in the past.[12]
There are currently no known environmental factors or triggers contributing to MCTD.[citation needed]It has been associated with HLA-DR4.[13]
Diagnosis
Distinguishing laboratory characteristics are a positive, speckled anti-nuclear antibody and an anti-U1-RNP antibody.[14][15]
After the original 1972 description of MCTD by Sharp, there was some controversy over whether MCTD was a distinct connective tissue disease, however after four decades and more than 2000 publications, it seems that there is a consensus that MCTD should be considered a distinctive clinical entity, and is thus considered as such by the majority of rheumatologists,[16] however there is a subgroup of patients that could evolve in their disease course towards another connective tissue disease.
Although almost any organ can be affected by MCTD, there are various clinical manifestations that make it more likely to suspect the disease is MCTD over other connective tissue diseases:[2]
- Raynaud’s phenomenon.
- Edematous hands and swollen fingers.
- Arthritis more severe than that of SLE.
- Pulmonary hypertension (does not need to be pulmonary fibrosis) differentiates MCTD from SLE and scleroderma.
- Anti-RNP antibodies in elevated levels, especially antibodies against protein 68 kD.
- Absence of severe renal or CNS disease.
Several criteria have been described to standardize the diagnosis of the disease, some of the most used being those of Alarcón-Segovia, although there are no universally accepted criteria.[17][18][19] In general the criteria require the presence of high titres of anti-RNP antibodies, the presence of some characteristic signs of the disease –Raynaud or swollen hands/fingers– and the presence of some clinical manifestations of at least two other connective tissue diseases –SLE, scleroderma, polymyositis.[citation needed]
| A. Serologic criteria:
Positive Anti-RNP at a titre> 1:160 by hemagglutination |
| B. Clinical Criteria
1. Edema of the hands 2. Synovitis 3. Myositis 4. Raynaud’s phenomenon 5. Acrosclerosis |
| MCTD is present with:
Criteria A together with 3 or more clinical criteria –one of which must be synovitis or myositis– |
It is often several years before sufficient signs and symptoms appear to make the diagnosis of MCTD, relative to the more sequential clinical manifestations of SLE, scleroderma, and polymyositis, so often, in the initial phases, the diagnosis most appropriate for patients is “undifferentiated connective tissue disease”.[19]
If the patient has edematous hands and/or swollen fingers in conjunction with elevated titers of antinuclear antibodies, an elevated titre of anti-U1 RNP antibody is a good predictor of progressing to MCTD.[20] The presence of this specific antibody is sine qua non for the diagnosis of MCTD,[19] although its isolated presence does not guarantee that a patient has MCTD or will develop it. If the dominant autoantibodies are antiDNAn, Sm, Scl70 or Ro, it is likely the patient will develop another connective disease distinct from MCTD. The clinical manifestations of MCTD appear correlated more intensely to the antibodies against protein A’ and 68 kD of the U1 RNP complex. The typical phenotype of MCTD also appears to be in part genetically determined, as patients with MCTD are associated with HLA-DR4 or HLA-DR2, meanwhile those with SLE are associated with HLA-DR3 and those with scleroderma are associated with HLA-DR5.[21]
SLE, scleroderma, and in MCTD have antibodies against anti-U1-snRNP at differing percentages. These antibodies are in most MCTD patients but are seen in only 30-35% of SLE and 2-14% of scleroderma patients, therefore they can help differentiate MCTD from other connective tissue disorders. There are different haplotypes of SNRNP70 which due to their differences in patients with MCTD versus those with SLE or scleroderma help substantiate the claim that MCTD is a separate disease. The T-G-CT-G haplotype is more common in patients with MCTD, whereas the T-G-C-G haplotype is more commonly seen in scleroderma and SLE.[22]
Differential diagnosis includes CPT2.
Treatment
Although MCTD was originally described as a disease with a good treatment response to corticosteroids, the treatment of the disease is based on the specific manifestations and clinical complications, similar to how other signs and symptoms are treated in other connective tissue disease. [23][24]
Standard
For arthritis, non-steroidal anti-inflammatories or low dose prednisone are usually used, which can be used in association with methotrexate or hydroxychloroquine. Temporomandibular joint arthritis has been shown to be successfully treated with condylar reconstruction using chondral grafts.[25] Higher doses of corticosteroids (0.25 to 1 mg/kg/day) are used in complications such as myositis, meningitis, pleuritis, pericarditis, myocarditis, interstitial lung disease, or hematologic abnormalities. On the contrary, Raynaud’s phenomenon, acrosclerosis or peripheral neuropathies are usually resistant to corticosteroids. Cyclophosphamide are useful in interstitial lung disease and in the eventual serious renal involvement. In cases of myositis or thrombocytopenias resistant to corticosteroids, intravenous immunoglobulins may be useful. For Raynaud, general measures (such as tobacco cessation, protection against the cold), calcium antagonists, endovenous prostaglandins or endothelin-2 antagonists may be useful. In patients with gastroesophageal reflux, proton pump inhibitors and H2 receptor antagonists can be used, following protocol for the usual treatment of these scleroderma problems.[23][24]
Since pulmonary hypertension is the leading cause of death, its early diagnosis by routine echocardiography and the rapid initiation of treatment with endothelin-1 antagonists (bosentan), phosphodiesterase 5 inhibitors (sildenafil) or endovenous prostacyclins (epoprostenol) manage to considerably improve morbidity and mortality.[23][24]
Investigational
Further investigation into appropriate treatment options for MCTD are in progress. Treatment for various rheumatoid diseases are currently undergoing research and have the potential to be used for patients presenting with similar signs and symptoms. Better understanding the pathophysiology of the disease and its progression will enable better targeted treatment options.[12]
Prognosis
The original description of the disease is characterized by a generally good prognosis and an excellent response to treatment with corticosteroids; however, in actuality it is clear that there is a group of patients with elevated morbidity and mortality. In a recent study the survival rates at 5, 10, and 15 years were 98%, 96%, and 88% respectively, with the main causes of death being pulmonary hypertension, cardiovascular problems, and infections.[26] The presence of anticardiolipin antibodies is a more serious risk factor for the disease, as well as the presence of more scleroderma and polymyositis signs and symptoms.[24]
Morbidity is quite high in patients with MCTD. In addition to fatigue and recurrent musculoskeletal complaints, patients can develop a fibromyalgia symptom as a result of occasional outbreaks requiring medium-high doses of corticosteroid. The steroids, in combination with their adverse effects, frequently cause fibromyalgia symptoms and thus complicate treatment.[24]
The prognosis of mixed connective tissue disease is in one third of cases worse than that of systemic lupus erythematosus (SLE). In spite of prednisone treatment, this disease is progressive and may in many cases evolve into a progressive systemic sclerosis (PSS), also referred to as diffuse cutaneous systemic scleroderma (dcSSc) which has a poor outcome. In some cases though the disease is mild and may only need aspirin as a treatment and may go into remission where no Anti-U1-RNP antibodies are detected, but that is rare or within 30% of cases.[citation needed] Most deaths from MCTD are due to heart failure caused by pulmonary arterial hypertension (PAH).
Disease progression
Patients diagnosed with MCTD may progress to a clinical picture more consistent with other connective tissue diseases like SLE, scleroderma, or rheumatoid arthritis. In some studies these patients become reclassified over time with other diseases, such as rheumatoid arthritis in 9%, SLE in 15%, and scleroderma in 21% of cases.[27] Such progression is, in part, determined genetically, thus SLE is more likely in patients with HLA-DR3 and scleroderma in patients with HLA-DR5.[24]
Epidemiology
The prevalence of MCTD is higher than that of dermatomyositis and lower than that of SLE.[28] In a 2011 Norwegian study, the prevalence of MCTD was 3.8 per 100,000 adults, with an incidence of 2.1 per million per year.[29]
MCTD is much more frequent in women than in men at between a 3:1 to 16:1 ratio, and in women younger than 50.[10] The general age at onset is around 15–25 years old.[citation needed]
See also
- Autoimmunity
- Overlap syndrome
- Rheumatoid arthritis
- Autoimmune disease
- Scleroderma
- Systemic lupus erythematosus
- Polymyositis
- Rheumatology
References
- ↑ 1.0 1.1 Rapini RP, Bolognia JL, Jorizzo JL (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 978-1-4160-2999-1.
- ↑ 2.0 2.1 2.2 Bennett, Robert (2014). "Clinical manifestations of mixed connective tissue disease". www.uptodate.com. Archived from the original on 2020-10-28. Retrieved 2019-10-12.
- ↑ 3.0 3.1 3.2 Ungprasert, Patompong; Crowson, Cynthia S.; Chowdhary, Vaidehi R.; Ernste, Floranne C.; Moder, Kevin G.; Matteson, Eric L. (December 2016). "Epidemiology of Mixed Connective Tissue Disease 1985-2014: A Population Based Study". Arthritis Care & Research. 68 (12): 1843–1848. doi:10.1002/acr.22872. ISSN 2151-464X. PMC 5426802. PMID 26946215.
- ↑ 4.0 4.1 Sharp GC, Irvin WS, Tan EM, Gould RG, Holman HR (February 1972). "Mixed connective tissue disease--an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA)". The American Journal of Medicine. 52 (2): 148–59. doi:10.1016/0002-9343(72)90064-2. PMID 4621694.
- ↑ Tsokos GC, Gordon C, Smolen JS (2007). Systemic lupus erythematosus: a companion to Rheumatology. Elsevier Health Sciences. pp. 429–. ISBN 978-0-323-04434-9.
- ↑ LeRoy EC, Maricq HR, Kahaleh MB (March 1980). "Undifferentiated connective tissue syndromes". Arthritis and Rheumatism. 23 (3): 341–3. doi:10.1002/art.1780230312. PMID 7362686.
- ↑ 7.0 7.1 Hoffman RW (1 June 2009). "Mixed Connective Disease". In Stone J (ed.). Pearls & Myths in Rheumatology. Springer. pp. 169–172. ISBN 978-1-84800-933-2. Retrieved 26 June 2010.
- ↑ "Mixed Connective Tissue Disease, MCTD". The Free Dictionary by Farlex. Archived from the original on 2018-07-13. Retrieved 2021-05-13.
- ↑ Nevares AM, Larner R. "Mixed Connective Tissue Disease (MCTD): Autoimmune Disorders of Connective Tissue". Merck Manual Home Health Handbook. Archived from the original on 2012-09-29. Retrieved 2021-05-13.
- ↑ 10.0 10.1 "Mixed connective tissue disease - Symptoms and causes". Mayo Clinic. Archived from the original on 2019-07-27. Retrieved 2019-10-12.
- ↑ Yang, Chia-Fu; Chiu, Jih-Yu; Su, Chang-Wei; Chen, Chun-Ming (2019). "Chondral grafts for condylar reconstruction in treatment of temporomandibular joint arthritis in a mixed connective tissue disease patient". The Kaohsiung Journal of Medical Sciences. 35 (12): 787–788. doi:10.1002/kjm2.12128. ISSN 2410-8650. PMID 31512336.
- ↑ 12.0 12.1 "Mixed Connective Tissue Disease (MCTD)". NORD (National Organization for Rare Disorders). Archived from the original on 2019-10-12. Retrieved 2019-10-12.
- ↑ Aringer M, Steiner G, Smolen JS (August 2005). "Does mixed connective tissue disease exist? Yes". Rheumatic Diseases Clinics of North America. 31 (3): 411–20, v. doi:10.1016/j.rdc.2005.04.007. PMID 16084315.
- ↑ Venables PJ (2006). "Mixed connective tissue disease". Lupus. 15 (3): 132–7. doi:10.1191/0961203306lu2283rr. PMID 16634365. S2CID 25736411. Archived from the original on 2021-08-28. Retrieved 2021-05-13.
- ↑ Sato T, Fujii T, Yokoyama T, Fujita Y, Imura Y, Yukawa N, Kawabata D, Nojima T, Ohmura K, Usui T, Mimori T (December 2010). "Anti-U1 RNP antibodies in cerebrospinal fluid are associated with central neuropsychiatric manifestations in systemic lupus erythematosus and mixed connective tissue disease". Arthritis and Rheumatism. 62 (12): 3730–40. doi:10.1002/art.27700. hdl:2433/142082. PMID 20722023.
- ↑ Cappelli, Susanna; Bellando Randone, Silvia; Martinović, Dušanka; Tamas, Maria-Magdalena; Pasalić, Katarina; Allanore, Yannick; Mosca, Marta; Talarico, Rosaria; Opris, Daniela; Kiss, Csaba G.; Tausche, Anne-Kathrin (February 2012). ""To be or not to be," ten years after: evidence for mixed connective tissue disease as a distinct entity". Seminars in Arthritis and Rheumatism. 41 (4): 589–598. doi:10.1016/j.semarthrit.2011.07.010. ISSN 1532-866X. PMID 21959290.
- ↑ Alarcón-Segovia, D.; Cardiel, M. H. (March 1989). "Comparison between 3 diagnostic criteria for mixed connective tissue disease. Study of 593 patients". The Journal of Rheumatology. 16 (3): 328–334. ISSN 0315-162X. PMID 2724251.
- ↑ "Sociedad Española de Reumatología :: Criterios diagnósticos". 2014-08-10. Archived from the original on 2014-08-10. Retrieved 2019-10-12.
- ↑ 19.0 19.1 19.2 Bennett, Robert. "Definition and diagnosis of mixed connective tissue disease". Archived from the original on 2020-08-15. Retrieved 2021-05-13.
- ↑ Greidinger, Eric L.; Hoffman, Robert W. (August 2005). "Autoantibodies in the pathogenesis of mixed connective tissue disease". Rheumatic Diseases Clinics of North America. 31 (3): 437–450, vi. doi:10.1016/j.rdc.2005.04.004. ISSN 0889-857X. PMID 16084317.
- ↑ Gendi, N. S.; Welsh, K. I.; Van Venrooij, W. J.; Vancheeswaran, R.; Gilroy, J.; Black, C. M. (February 1995). "HLA type as a predictor of mixed connective tissue disease differentiation. Ten-year clinical and immunogenetic followup of 46 patients". Arthritis and Rheumatism. 38 (2): 259–266. doi:10.1002/art.1780380216. ISSN 0004-3591. PMID 7848317.
- ↑ "Table 1: The Single Nucleotide Polymorphisms in cathepsin B protein mined from literature (PMID: 16492714)". doi:10.7717/peerj.7425/table-1.
{{cite journal}}: Cite journal requires|journal=(help) - ↑ 23.0 23.1 23.2 Mobasat, A.; Turrión Nieves, A.; Bohorquez Heras, C. (January 2013). "Enfermedad mixta del tejido conectivo. Síndromes de solapamiento". Medicine - Programa de Formación Médica Continuada Acreditado. 11 (32): 1991–1996. doi:10.1016/s0304-5412(13)70567-5. ISSN 0304-5412.
- ↑ 24.0 24.1 24.2 24.3 24.4 24.5 Bennett, Robert. "Prognosis and treatment of mixed connective tissue disease". Archived from the original on 2020-08-06. Retrieved 2021-05-13.
- ↑ Yang, Chia‐Fu; Chiu, Jih‐Yu; Su, Chang‐Wei; Chen, Chun‐Ming (2019-09-11). "Chondral grafts for condylar reconstruction in treatment of temporomandibular joint arthritis in a mixed connective tissue disease patient". The Kaohsiung Journal of Medical Sciences. 35 (12): 787–788. doi:10.1002/kjm2.12128. ISSN 1607-551X. PMID 31512336.
- ↑ Hajas, Agota; Szodoray, Peter; Nakken, Britt; Gaal, Janos; Zöld, Eva; Laczik, Renata; Demeter, Nora; Nagy, Gabor; Szekanecz, Zoltan; Zeher, Margit; Szegedi, Gyula (July 2013). "Clinical course, prognosis, and causes of death in mixed connective tissue disease". The Journal of Rheumatology. 40 (7): 1134–1142. doi:10.3899/jrheum.121272. ISSN 0315-162X. PMID 23637328. S2CID 19625256.
- ↑ Ruiz Pombo, Mónica; Labrador Horrillo, Moisés; Selva O'Callaghan, Albert (2004-11-20). "[Undifferentiated, overlapping and mixed connective tissue diseases]". Medicina Clinica. 123 (18): 712–717. doi:10.1016/s0025-7753(04)75337-3. ISSN 0025-7753. PMID 15563821.
- ↑ "Mixed Connective-Tissue Disease: Practice Essentials, Pathophysiology, Etiology". 2019-07-17. Archived from the original on 2021-05-07. Retrieved 2021-05-13.
{{cite journal}}: Cite journal requires|journal=(help) - ↑ Gunnarsson, Ragnar; Molberg, Oyvind; Gilboe, Inge-Margrethe; Gran, Jan Tore; PAHNOR1 Study Group (June 2011). "The prevalence and incidence of mixed connective tissue disease: a national multicentre survey of Norwegian patients". Annals of the Rheumatic Diseases. 70 (6): 1047–1051. doi:10.1136/ard.2010.143792. hdl:10852/36041. ISSN 1468-2060. PMID 21398332. S2CID 206864939.
Further reading
- Alana M. Nevares, Mixed Connective Tissue Disease (MCTD), MSD Manual: Consumer edition (April 2018) Archived 2020-09-19 at the Wayback Machine, Professional edition (February 2018) Archived 2020-07-30 at the Wayback Machine
- Shiel WC (May 2016). Driver CB (ed.). "Mixed Connective Tissue Disease (MCTD)". MedicineNet.com. Archived from the original on 2021-04-21. Retrieved 2021-05-13.
- Nevares AM, Larner R. "Mixed Connective Tissue Disease (MCTD): Autoimmune Rheumatic Disorders". Merck Manual Professional. Archived from the original on 2015-03-21. Retrieved 2021-05-13.
- Racaza G, Gonzales Penserga E (January–March 2014). "Mixed Connective Tissue Disease in Filipinos – A 13-Year Retrospective Review of 14 Cases in the Philippine General Hospital". Philippine Journal of Internal Medicine. 52 (1): 1–7. Archived from the original on 2021-08-28. Retrieved 2021-05-13.
External links
| Classification | |
|---|---|
| External resources |
- Pages with script errors
- CS1 errors: missing periodical
- All articles with unsourced statements
- Articles with unsourced statements from October 2020
- Articles with invalid date parameter in template
- Articles with unsourced statements from January 2017
- Articles with unsourced statements from November 2019
- Webarchive template wayback links
- Connective tissue diseases
- Disorders of fascia
- Systemic connective tissue disorders
- Autoimmune diseases