Cholesteatoma
| Cholesteatoma | |
|---|---|
| Other names: Keratomas[1] | |
 | |
| Cholesteatoma | |
| Specialty | ENT surgery |
| Symptoms | Drainage from the ear, ear pain[1] |
| Complications | Hearing loss, ringing in the ears[1] |
| Duration | Long-term[2] |
| Types | Congenital, acquired[1] |
| Risk factors | Family history, otitis media, injury to the ear[1] |
| Diagnostic method | Physical examination[2] |
| Differential diagnosis | Tympanosclerosis; middle ear osteoma; chronic suppurative otitis media[1][2] |
| Treatment | Surgical removal[1] |
| Frequency | 3 per 100,000 children/year[1] 9 per 100,000 adults/year[1] |
Cholesteatoma is an expanding and destructive growth of the middle ear or mastoid.[1] Symptoms may include drainage from the ear with or without ear pain.[1][2] This may be long-term in nature.[2] Complications can include hearing loss, ringing in the ears, and infection.[1][2]
Risk factors include family history of the condition, otitis media, and injury to the ear.[1] People with cleft palate, Turner syndrome, and Down syndrome are more commonly affected.[1] They consist of keratin and squamous epithelium.[1] They are not a cancer.[1] Diagnosis is based on the appearance and may be supported by medical imaging.[2]
Treatment generally consists of surgical removal.[1] Surgery; however, may not restore prior hearing loss.[2] Additionally between 5% and 50% of cases recur following removal and repeat surgery may be required.[2]
Cholesteatoma occur in about 3 per 100,000 children and 9 per 100,000 adults a year.[1] Males are affected more often than females.[1] The condition was first described in 1683 by Du Verney.[1] It was named in 1838 by Johannes Müller.[1] The term is from "chole" meaning "cholesterol", "steat" meaning "fat", and "oma" meaning "tumor"; though the lesion is not from fat tissue.[1]
Signs and symptoms
Looking in the ear may only reveal a canal full of discharge. Only once the ear is cleaned and the entire tympanic membrane is inspected can a cholesteatoma be diagnosed.
Once the debris is cleared, cholesteatoma can give rise to a number of appearances. If there is significant inflammation, the tympanic membrane may be partially obscured by an aural polyp. If there is less inflammation, the cholesteatoma may present the appearance of 'semolina' discharging from a defect in the tympanic membrane. The posterior and superior parts of the tympanic membrane are most commonly affected. If the cholesteatoma has been dry, the cholesteatoma may present the appearance of 'wax over the attic'. The attic is just above the eardrum.
Other less common symptoms (all less than 15%) may include pain, balance disruption, tinnitus, earache, headaches and bleeding from the ear. There can also be facial nerve weakness. Balance symptoms raise the possibility that the cholesteatoma is eroding the balance organs in the inner ear.
If untreated, a cholesteatoma can eat into the three small bones located in the middle ear (the malleus, incus and stapes, collectively called ossicles), which can result in nerve deterioration, deafness, imbalance and vertigo. It can also affect and erode the thin bone structure that isolates the top of the ear from the brain, which increase the risk of brain infection including brain abscess.
Both the acquired as well as the congenital types of the disease can affect the facial nerve that extends from the brain to the face and passes through the inner and middle ear and leaves at the anterior tip of the mastoid bone, and then rises to the front of the ear and extends into the upper and lower face.
-
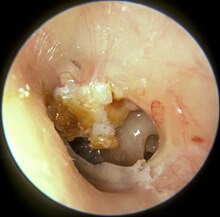
Cholesteatoma and large perforation left ear drum
-
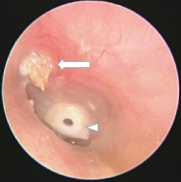
Acquired cholesteatoma of attic in left ear

.png)
Cause
Cholesteatomas occur in two basic classifications: Acquired cholesteatomas, which are more common, are usually caused by pathological alteration of the ear drum leading to accumulation of keratin within the middle ear. Congenital cholesteatomas are usually middle ear epidermal cysts that are identified deep within an intact ear drum.
Congenital
Keratin-filled cysts that grow medial to the tympanic membrane are considered to be congenital if they fulfill the following criteria (Levenson's criteria):[3]
- mass medial to the tympanic membrane
- normal tympanic membrane
- no previous history of ear discharge, perforation or ear surgery
Congenital cholesteatomas occur at three important sites: the middle ear, the Petrous apex, and the cerebropontinio angle. They are most often found deep to the anterior aspect of the ear drum, and a vestigial structure, the epidermoid formation, from which congenital cholesteatoma may originate, has been identified in this area.[4]
Not all middle ear epidermal cysts are congenital, as they can be acquired either by metaplasia of the middle ear mucosa or by traumatic implantation of ear canal or tympanic membrane skin. In addition, cholesteatoma inadvertently left by a surgeon usually regrows as an epidermal cyst. Some authors have also suggested hereditary factors.[5][6]
Acquired
More commonly, keratin accumulates in a pouch of tympanic membrane which extends into the middle ear space. This abnormal folding or 'retraction' of the tympanic membrane arises in one of the following ways:
- Jackler’s theory : Mucosal coupling with traction generated by interaction of migrating opposing surfaces leading to formation of cholesteatoma.[7]
- Wittmaack's theory : Invagination of tympanic membrane from the attic or part of pars tensa in the form of retraction pockets lead to the formation of cholesteatoma.[8]
- Ruedi's theory : The basal cells of germinal layer of skin proliferate under the influence of infection and lay down keratinising squamous epithelium.[9]
- Habermann's theory: The epithelium from the meatus or outer drum surface grows into the middle ear through a pre-existing perforation and form cholesteatoma.[10]
Cholesteatoma may also arise as a result of metaplasia of the middle ear mucosa [11] or implantation following trauma.
Diagnosis
Cholesteatoma is diagnosed by physical examination of the ear. A CT scan may help to rule out other, often more serious causes for the person's symptoms. MRI may used aswell.[12][13]
-

Cholesteatoma as seen during microscopic examination
-
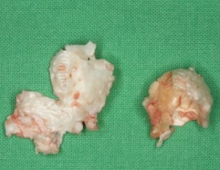
Dissected friable cholesteatoma with a thin pearly-white greasy-looking wall containing pultaceous substance

.png)
Differential diagnosis
Other more common conditions (e.g. otitis externa) may present with these symptoms, but cholesteatoma is more serious and should not be overlooked. If a people presents to a doctor with ear discharge and hearing loss, the doctor should consider cholesteatoma.
Treatment
Cholesteatoma is a persistent disease. Once the diagnosis is made in a patient who can tolerate a general anesthetic, the standard treatment is to surgically remove the growth.
The challenge of cholesteatoma surgery is to permanently remove the cholesteatoma whilst retaining or reconstructing the normal functions of the structures housed within the temporal bone.
The general objective of cholesteatoma surgery has two parts. It is both directed against the underlying pathology and directed towards maintaining the normal functions of the temporal bone. These aims are conflicting and this makes cholesteatoma surgery extremely challenging.
Sometimes, the situation results in a clash of surgical aims. The need to fully remove a progressive disease like cholesteatoma is the surgeon's first priority. Preservation of hearing is secondary to this primary aim. If the disease can be removed easily so that there is no increased risk of residual disease, then the ossicles may be preserved. If the disease is difficult to remove, so that there is an increased risk of residual disease, then removal of involved ossicles in order to fully clear cholesteatoma has generally been regarded as necessary and reasonable.
In other words, the aims of cholesteatoma treatment form a hierarchy. The paramount objective is the complete removal of cholesteatoma. The remaining objectives, such as hearing preservation, are subordinate to the need for complete removal of cholesteatoma. This hierarchy of aims has led to the development of a wide range of strategies for the treatment of cholesteatoma.
Surgery
The variation in technique in cholesteatoma surgery results from each surgeon's judgment whether to retain or remove certain structures housed within the temporal bone in order to facilitate the removal of cholesteatoma. This typically involves some form of mastoidectomy which may or may not involve removing the posterior ear canal wall and the ossicles.
Removal of the canal wall facilitates the complete clearance of cholesteatoma from the temporal bone in three ways:
- It removes a large surface onto which cholesteatoma may be adherent;
- It removes a barrier behind which the cholesteatoma may be hidden;
- It removes an impediment to the introduction of instruments used for the removal of cholesteatoma.
Thus removal of the canal wall provides one of the most effective strategies for achieving the primary aim of cholesteatoma surgery, the complete removal of cholesteatoma. However, there is a trade-off, since the functional impact of canal wall removal is also important.
The removal of the ear canal wall results in:
- a space, the "mastoid cavity", which is less likely than the original ear canal to resist infection;
- exposure of the ossicles, which may allow the subsequent formation of a new cholesteatoma deep to the ossicles. To prevent this, these ossicles must be removed, which may diminish the patient's hearing.
The formation of a mastoid cavity by removal of the canal wall is the simplest and most effective procedure for facilitating the removal of cholesteatoma, but may bestow the most lasting infirmity due to loss of ear function upon the patient treated in this way.
The following strategies are employed to mitigate the effects of canal wall removal:
- Careful design and construction of the mastoid cavity. This is essential for the health and integrity of the protective sheet of migrating, keratising epithelium which lines the distorted ear canal. This requires the surgeon to saucerise the cavity. A high facial ridge and an inappropriately small cartilaginous meatus are obstructions to epithelial migration and are particularly high risk factors for failure of the self-cleaning mechanism of the external ear.[14]
- Partial obliteration of the mastoid cavity. This can be performed using a wide range of materials. Many of these resorb in time, which means that the long-term results of such surgery are poorer than the short-term results.[15]
- Reconstruction of the ear canal wall. Canal wall reconstruction has been performed using ear canal skin alone, fascia, cartilage, titanium as well as by replacing the original intact wall. If the reconstruction is poorly performed, it may result in a high rate of recurrent cholesteatoma.[16]
- Preservation of the ear canal wall. If poorly performed, it may result in a high rate of both residual and recurrent cholesteatoma.[17]
- Reconstruction of the chain of hearing bones using a passive middle ear implant.[18]
Clearly, preservation and restoration of ear function at the same time as total removal of cholesteatoma requires a high level of surgical expertise.
Endoscopic

Traditionally, ear surgery has been performed using the surgical microscope. The direct line of view dictated by that approach necessitates using the mastoid as the access port to the middle ear. It has long been recognized that failure in cholesteatoma surgery occurs in some of the out of view spaces of the tympanic cavity like the sinus tympani and facial recess that are out of view using the traditional microscopic technique.[19] More recently, the endoscope has been increasingly utilized in the surgical management of cholesteatoma in one of two ways:
- Using the endoscope as an ancillary instrument to the microscope when trying to visualize areas that are hidden from the microscope such as the sinus tympani. This work was pioneered by Professor Thomassin.[20]
- Using the endoscope as the main surgical instrument through the ear canal, or what's called: endoscopic ear surgery.[21]
There are multiple advantages for the use of the endoscope in cholesteatoma surgery:
- The endoscope wide angle of view and the ability to "see around the corner".[22]
- The endoscope allows minimally invasive access through the natural ear canal, rather than through the usual 5 cm incision behind the ear utilized in traditional microscopic surgery.[22]
- The 30 degrees endoscope allows access to the bony area of the Eustachian tube to address any obstructive pathologies.[22]
Prognosis
It is important that the patient attend periodic follow-up checks, because even after careful microscopic surgical removal, cholesteatomas may recur. Such recurrence may arise many years, or even decades, after treatment.
A residual cholesteatoma may develop if the initial surgery failed to completely remove the original; residual cholesteatomas typically become evident within the first few years after the initial surgery.
A recurrent cholesteatoma is a new cholesteatoma that develops when the underlying causes of the initial cholesteatoma are still present. Such causes can include, for example, poor Eustachian tube function, which results in retraction of the ear drum, and failure of the normal outward migration of skin.[23]
In a retrospective study of 345 patients with middle ear cholesteatoma operated on by the same surgeon, the overall 5-year recurrence rate was 11.8%.[24] In a different study with a mean follow-up period of 7.3 years, the recurrence rate was 12.3%, with the recurrence rate being higher in children than in adults.[25] The use of the endoscope as an ancillary instrument has been shown to reduce the incidence of residual cholesteatoma.[26] Although more studies are needed, so far, new techniques addressing underlying Eustachian tube dysfunction such as transtympanic dilatation of the Eustachian tube has not been shown to change outcomes of chronic ear surgery.[27]
Epidemiology
The number of new cases of cholesteatoma in Iowa was estimated in 1975–76 to be just under one new case per 10,000 citizens per year.[28] Cholesteatoma affects all age groups, from infants through to the elderly. The peak incidence occurs in the second decade.[28]
See also
References
- ↑ 1.00 1.01 1.02 1.03 1.04 1.05 1.06 1.07 1.08 1.09 1.10 1.11 1.12 1.13 1.14 1.15 1.16 1.17 1.18 1.19 1.20 1.21 Castle, JT (September 2018). "Cholesteatoma Pearls: Practical Points and Update". Head and neck pathology. 12 (3): 419–429. doi:10.1007/s12105-018-0915-5. PMID 30069838.
- ↑ 2.0 2.1 2.2 2.3 2.4 2.5 2.6 2.7 2.8 Kennedy, Kenneth L.; Singh, Achint K. (2023). "Middle Ear Cholesteatoma". StatPearls. StatPearls Publishing. Archived from the original on 21 October 2022. Retrieved 13 August 2023.
- ↑ Derlacki EL, Clemis JD (September 1965). "Congenital cholesteatoma of the middle ear and mastoid". The Annals of Otology, Rhinology, and Laryngology. 74 (3): 706–727. doi:10.1177/000348946507400313. PMID 5846535. S2CID 26714025.
- ↑ Michaels L (February 1988). "Origin of congenital cholesteatoma from a normally occurring epidermoid rest in the developing middle ear". International Journal of Pediatric Otorhinolaryngology. 15 (1): 51–65. doi:10.1016/0165-5876(88)90050-X. PMID 3286554.
- ↑ Lipkin AF, Coker NJ, Jenkins HA (October 1986). "Hereditary congenital cholesteatoma. A variant of branchio-oto dysplasia". Archives of Otolaryngology–Head & Neck Surgery. 112 (10): 1097–1100. doi:10.1001/archotol.1986.03780100085014. PMID 3755982.
- ↑ Landegger LD, Cohen MS (November 2013). "Congenital cholesteatoma in siblings". The Journal of Laryngology and Otology. 127 (11): 1143–1144. doi:10.1017/S0022215113002284. PMID 24169145.
- ↑ Jackler, Robert K.; Santa Maria, Peter L.; Varsak, Yasin K.; Nguyen, Anh; Blevins, Nikolas H. (August 2016). "A new theory on the pathogenesis of acquired cholesteatoma: Mucosal traction". The Laryngoscope. 125 Suppl 4: S1–S14. doi:10.1002/lary.25261. ISSN 1531-4995. PMID 26013635. Archived from the original on 2022-09-03. Retrieved 2022-09-24.
- ↑ Becvarovski Z. "Chronic suppurative otitis media". ENT. Archived from the original on 18 January 2013. Retrieved 12 January 2013.
- ↑ Ruedi L (1959). "Cholesteatoma formation in the middle ear in animal experiments". Acta Oto-Laryngologica. 50 (3–4): 233–240, discussion 240–242. doi:10.3109/00016485909129191. PMID 13660782.
- ↑ Haberman J (1888). "Zur Entstehung des Cholesteatoms des Mittelohrs". Archiv für Ohrenheilkunde. 27 (2–3): 43–51. doi:10.1007/BF02104525.
- ↑ Sadé J, Babiacki A, Pinkus G (1983). "The metaplastic and congenital origin of cholesteatoma". Acta Oto-Laryngologica. 96 (1–2): 119–129. doi:10.3109/00016488309132882. PMID 6193677.
- ↑ Patel VA, Isildak H, Khaku AM (2018-09-17). Meyers AD (ed.). "Cholesteatoma: Practice Essentials, Background, Etiology and Pathophysiology". Medscape. Archived from the original on 2022-08-30. Retrieved 2022-09-24.
- ↑ "Cholesteatoma". MedlinePlus Medical Encyclopedia. U.S. National Library of Medicine. Archived from the original on 2021-03-19. Retrieved 2018-09-21.
- ↑ Wormald PJ, Nilssen EL (January 1998). "The facial ridge and the discharging mastoid cavity". The Laryngoscope. 108 (1 Pt 1): 92–96. doi:10.1097/00005537-199801000-00017. PMID 9432074. S2CID 21562748.
- ↑ Black B (December 1995). "Mastoidectomy elimination". The Laryngoscope. 105 (12 Pt 2 Suppl 76): 1–30. doi:10.1288/00005537-199512000-00023. PMID 7500804.
- ↑ Deveze A, Rameh C, Puchol MS, Lafont B, Lavieille JP, Magnan J (February 2010). "Rehabilitation of canal wall down mastoidectomy using a titanium ear canal implant". Otology & Neurotology. 31 (2): 220–224. doi:10.1097/MAO.0b013e3181c9960d. PMID 20009781. S2CID 23723605.
- ↑ Jansen C (September 1968). "The combined approach for tympanoplasty (report on 10 years' experience)". The Journal of Laryngology and Otology. 82 (9): 779–793. doi:10.1017/S0022215100069462. PMID 4878658.
- ↑ Austin DF (December 1971). "Ossicular reconstruction". Archives of Otolaryngology. 94 (6): 525–535. doi:10.1001/archotol.1971.00770070825007. PMID 5129224.
- ↑ Sheehy JL, Brackmann DE, Graham MD (July 1977). "Cholesteatoma surgery: residual and recurrent disease. A review of 1,024 cases". The Annals of Otology, Rhinology, and Laryngology. 86 (4 Pt 1): 451–462. doi:10.1177/000348947708600405. PMID 889222.
- ↑ Thomassin JM, Korchia D, Doris JM (August 1993). "Endoscopic-guided otosurgery in the prevention of residual cholesteatomas". The Laryngoscope. 103 (8): 939–943. doi:10.1288/00005537-199308000-00021. PMID 8361301.
- ↑ Tarabichi M (January 1999). "Endoscopic middle ear surgery". The Annals of Otology, Rhinology, and Laryngology. 108 (1): 39–46. doi:10.1177/000348949910800106. PMID 9930539.
- ↑ 22.0 22.1 22.2 Kapadiya M, Tarabichi M (June 2019). "An overview of endoscopic ear surgery in 2018". Laryngoscope Investigative Otolaryngology. 4 (3): 365–373. doi:10.1002/lio2.276. PMC 6580051. PMID 31236473.
- ↑ Fairley J (7 November 2010). "Cholesteatoma and mastoid surgery". entkent.com. Archived from the original on 13 December 2012. Retrieved 29 December 2012.
- ↑ Mishiro Y, Sakagami M, Kitahara T, Kondoh K, Okumura S (September 2008). "The investigation of the recurrence rate of cholesteatoma using Kaplan-Meier survival analysis". Otology & Neurotology. 29 (6): 803–806. doi:10.1097/MAO.0b013e318181337f. PMID 18636031. S2CID 25970515.
- ↑ Vartiainen E (July 1995). "Factors associated with recurrence of cholesteatoma". The Journal of Laryngology and Otology. 109 (7): 590–592. doi:10.1017/S0022215100130804. PMID 7561462.
- ↑ Badr-el-Dine M (September 2002). "Value of ear endoscopy in cholesteatoma surgery". Otology & Neurotology. 23 (5): 631–635. doi:10.1097/00129492-200209000-00004. PMID 12218610.
- ↑ Kapadia M, Tarabichi M (October 2018). "Feasibility and Safety of Transtympanic Balloon Dilatation of Eustachian Tube". Otology & Neurotology. 39 (9): e825–e830. doi:10.1097/MAO.0000000000001950. PMID 30124616.
- ↑ 28.0 28.1 Harker LA (1977). Cholesteatoma: an incidence study in Cholesteatoma First International Conference. Birmingham, Alabama: Aesculapius Publishing Company. pp. 308–309. ISBN 978-0-912684-11-6.
External links
| Classification | |
|---|---|
| External resources |
- Information on Cholesteatomas Archived 2014-07-28 at the Wayback Machine
- Laser Cholesteatoma Surgery Archived 2022-03-16 at the Wayback Machine