Neuropathy, ataxia, and retinitis pigmentosa
| Neuropathy, ataxia, and retinitis pigmentosa | |
|---|---|
| Other names: Neurogenic muscle weakness-ataxia-retinitis pigmentosa syndrome | |
 | |
| This condition is inherited via a mitochondrial inheritance manner | |
Neuropathy, ataxia, and retinitis pigmentosa, also known as NARP syndrome, is a rare disease with mitochondrial inheritance that causes a variety of signs and symptoms chiefly affecting the nervous system[1] Beginning in childhood or early adulthood, most people with NARP experience numbness, tingling, or pain in the arms and legs (sensory neuropathy); muscle weakness; and problems with balance and coordination (ataxia). Many affected individuals also have vision loss caused by changes in the light-sensitive tissue that lines the back of the eye (the retina).[2][3] In some cases, the vision loss results from a condition called retinitis pigmentosa. This eye disease causes the light-sensing cells of the retina gradually to deteriorate.
Signs and symptoms
Learning disabilities and developmental delays are often seen in children with NARP, and older individuals with this condition may experience a loss of intellectual function (dementia). Other features of NARP include seizures, hearing loss, and abnormalities of the electrical signals that control the heartbeat (cardiac conduction defects).[4] These signs and symptoms vary among affected individuals.[citation needed]
Genetics
Neuropathy, ataxia, and retinitis pigmentosa is a condition related to changes in mitochondrial DNA. Mutations in the MT-ATP6 gene cause neuropathy, ataxia, and retinitis pigmentosa.[5] The MT-ATP6 gene provides instructions for making a protein that is essential for normal mitochondrial function. Through a series of chemical reactions, mitochondria use oxygen and simple sugars to create adenosine triphosphate (ATP), the cell's main energy source. The MT-ATP6 protein forms one part (subunit) of an enzyme called ATP synthase, which is responsible for the last step in ATP production.[6] Mutations in the MT-ATP6 gene alter the structure or function of ATP synthase, reducing the ability of mitochondria to make ATP.[7] It remains unclear how this disruption in mitochondrial energy production leads to muscle weakness, vision loss, and the other specific features of NARP.[citation needed]
This condition is inherited in a pattern reflecting its location in mitochondrial DNA, which is also known as maternal inheritance. This pattern of inheritance applies to genes contained in mitochondrial DNA. Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, only females pass mitochondrial conditions to their children. Mitochondrial disorders can appear in every generation of a family and can affect both males and females, but fathers do not pass mitochondrial traits to their children. Most of the body's cells contain thousands of mitochondria, each with one or more copies of mitochondrial DNA. The severity of some mitochondrial disorders is associated with the percentage of mitochondria in each cell that has a particular genetic change. Most individuals with NARP have a specific MT-ATP6 mutation in 70 percent to 90 percent of their mitochondria. When this mutation is present in a higher percentage of a person's mitochondria—greater than 90 percent to 95 percent—it causes a more severe condition known as maternally inherited Leigh syndrome. Because these two conditions result from the same genetic changes and can occur in different members of a single family, researchers believe that they may represent a spectrum of overlapping features instead of two distinct syndromes.[citation needed]
Diagnosis
The clinical diagnosis is backed up by investigative findings. Citrulline level in blood is decreased.[7][8] Mitochondrial studies or NARP mtDNA evaluation plays a role in genetic diagnosis[9] which can also be done prenatally.[10] The following, though not exaustive, are some other evaluation methods:[11]
- MRI
- Electromyography
-
![Hyperautofluorescence and hypoautofluorescence granular patterns in posterior pole[12]](https://nccommons.org/media/thumb/d/d6/Cabr-15-486-g002.jpg/371px-Cabr-15-486-g002.jpg)
Hyperautofluorescence and hypoautofluorescence granular patterns in posterior pole[12]
-
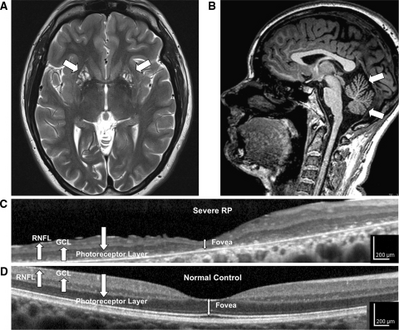
a-d)High-resolution retinal and brain imaging in NARP syndrome demonstrates analogous patterns of tissue injury
![Hyperautofluorescence and hypoautofluorescence granular patterns in posterior pole[12]](/wiki/File:Cabr-15-486-g002.jpg)

Treatment
There is currently no known cure for NARP syndrome. Symptomatic relief is targeted. Antioxidants play a role in improving the oxidative phosphorylation that is otherwise impaired.[13]
Prognosis
The severity and prognosis vary with the type of mutation involved.[14]
See also
References
- ↑ "Maternally inherited Leigh syndrome and NARP syndrome". rarediseases.org. NORD's Rare Disease Information Database. Archived from the original on 27 September 2014. Retrieved 27 September 2014.
- ↑ Kerrison, JB; Biousse, V; Newman, NJ (Feb 2000). "Retinopathy of NARP syndrome". Archives of Ophthalmology. 118 (2): 298–9. doi:10.1001/archopht.118.2.298. PMID 10676807.
- ↑ Chowers, I; Lerman-Sagie, T; Elpeleg, ON; Shaag, A; Merin, S (Feb 1999). "Cone and rod dysfunction in the NARP syndrome". The British Journal of Ophthalmology. 83 (2): 190–3. doi:10.1136/bjo.83.2.190. PMC 1722923. PMID 10396197.
- ↑ Keränen, T; Kuusisto, H (Sep 2006). "NARP syndrome and adult-onset generalised seizures". Epileptic Disorders. 8 (3): 200–3. PMID 16987741. Archived from the original on 2016-09-20. Retrieved 2022-06-06.
- ↑ Thorburn, DR; Rahman, S; Pagon, RA; Adam, MP; Ardinger, HH; Bird, TD; Dolan, CR; Fong, CT; Smith, RJH; Stephens, K (1993). Mitochondrial DNA-Associated Leigh Syndrome and NARP. University of Washington, Seattle. PMID 20301352. Archived from the original on 2017-01-28. Retrieved 2022-06-06.
- ↑ Rak, M; Tetaud, E; Duvezin-Caubet, S; Ezkurdia, N; Bietenhader, M; Rytka, J; di Rago, JP (Nov 23, 2007). "A yeast model of the neurogenic ataxia retinitis pigmentosa (NARP) T8993G mutation in the mitochondrial ATP synthase-6 gene". The Journal of Biological Chemistry. 282 (47): 34039–47. doi:10.1074/jbc.M703053200. PMID 17855363.
- ↑ 7.0 7.1 Parfait, B; de Lonlay, P; von Kleist-Retzow, JC; Cormier-Daire, V; Chrétien, D; Rötig, A; Rabier, D; Saudubray, JM; Rustin, P; Munnich, A (Jan 1999). "The neurogenic weakness, ataxia and retinitis pigmentosa (NARP) syndrome mtDNA mutation (T8993G) triggers muscle ATPase deficiency and hypocitrullinaemia". European Journal of Pediatrics. 158 (1): 55–8. doi:10.1007/s004310051009. PMID 9950309. S2CID 42374353.
- ↑ "NARP syndrome". metagene.de. MIC - Metabolic Information Centre. Archived from the original on 4 March 2016. Retrieved 27 September 2014.
- ↑ "ARP mtDNA Evaluation". athenadiagnostics.com. Athena Diagnostics, Inc. Archived from the original on 7 November 2014. Retrieved 27 September 2014.
- ↑ "Mitochondrial Studies: NARP - Neuropathy, Ataxia and Retinitis Pigmentosa". knightdxlabs.com. Archived from the original on 5 March 2016. Retrieved 27 September 2014.
- ↑ "Orphanet: NARP syndrome". www.orpha.net. Archived from the original on 2024-04-18. Retrieved 2024-04-17.
- ↑ Juaristi, Leire; Irigoyen, Cristina; Quiroga, Jorge (July 2021). "NEUROPATHY, ATAXIA, AND RETINITIS PIGMENTOSA SYNDROME: A MULTIDISCIPLINARY DIAGNOSIS". RETINAL Cases & Brief Reports. 15 (4): 486–489. doi:10.1097/ICB.0000000000000835. Archived from the original on 2024-04-18. Retrieved 2024-04-17.
- ↑ Mattiazzi, M; Vijayvergiya, C; Gajewski, CD; DeVivo, DC; Lenaz, G; Wiedmann, M; Manfredi, G (Apr 15, 2004). "The mtDNA T8993G (NARP) mutation results in an impairment of oxidative phosphorylation that can be improved by antioxidants". Human Molecular Genetics. 13 (8): 869–79. doi:10.1093/hmg/ddh103. PMID 14998933.
- ↑ Debray, FG; Lambert, M; Lortie, A; Vanasse, M; Mitchell, GA (Sep 1, 2007). "Long-term outcome of Leigh syndrome caused by the NARP-T8993C mtDNA mutation". American Journal of Medical Genetics Part A. 143A (17): 2046–51. doi:10.1002/ajmg.a.31880. PMID 17663470. S2CID 25756985.
Further reading
- Neuropathy, ataxia, and retinitis pigmentosa at NLM Genetics Home Reference
- NARP syndrome:ORPHA644 Archived 2016-03-04 at the Wayback Machine on orpha.net
- Nyhan, William L; Barshop, Bruce A; Ozand, Pinar T. (26 August 2005). Atlas of Metabolic Diseases Second edition. CRC Press. p. 350. ISBN 978-0-340-80970-9.
External links
| Classification | |
|---|---|
| External resources |