X-linked agammaglobulinemia
| X-linked agammaglobulinemia | |
|---|---|
| Other names: Bruton agammaglobulinemia, Btk agammaglobulinemia, Bruton tyrosine kinase agammaglobulinemia, agammaglobulinemia of Bruton, congenital agammaglobulinemia, X-linked hypogammaglobulinemia, Bruton type agammaglobulinemia, Bruton syndrome, sex-linked agammaglobulinemia[1][2] | |
 | |
| The disorder is passed on in an X-linked recessive pattern | |
| Specialty | Medical genetics |
| Symptoms | No tonsils, recurrent bacterial infections[3][4] |
| Complications | Bronchiectasis, cancer[4] |
| Usual onset | 6 months of age[3] |
| Causes | Mutation of the Btk gene[3] |
| Diagnostic method | Low immunoglobulins and B cells, genetic testing[4] |
| Differential diagnosis | Autosomal recessive agammaglobulinemia, autosomal dominant agammaglobulinemia, common variable immunodeficiency, hyper IgM syndrome, combined immunodeficiency[3] |
| Treatment | Immunoglobulin therapy, antibiotics, vaccination[3] |
| Frequency | 1 in 100,000 newborn males[1] |
X-linked agammaglobulinemia (XLA) is a genetic disorder that affects the body's ability to fight infection.[1] Those affected typically present around 6 months of age with recurrent bacterial infections including pneumonia, middle ear infections, cellulitis, diarrhea, and conjunctivitis.[3] Other symptoms include a lack of tonsils and no lymph nodes.[4] Complications can include bronchiectasis and cancer.[4]
Its is caused by various mutations of the Bruton's tyrosine kinase (Btk) gene and is inherited in an X-linked recessive manner.[3][1] The underlying mechanism involves an inability to form mature B cells, and thus the inability to make antibodies.[4] It is a type of primary immunodeficiency disorder.[4] Diagnosis is usually based on low immunoglobulins and B cells in the blood and can be confirmed by genetic testing.[4]
There is no cure.[1] Treatment is generally with immunoglobulin therapy either through a vein (IVIG) or subcutaneously (SCIG).[3] When infections do occur, longer courses of antibiotics are generally required.[3] Long-term antibiotics may also be used to try to prevent infections.[3] Those affected should receive all vaccines except live vaccines.[3][1] Treatment is required for survival and with treatment survival is generally beyond the age of 40.[1]
XLA affects about 1 in 100,000 live newborn males.[1] It was described by Ogden Bruton in 1952 and was the first known primary immunodeficiency disorder.[5][6] Effective treatment became available in the 1970s; however, is often costly.[1]
Signs and symptoms

Affects males 50% of the time if mother is a carrier for the gene. Children are generally asymptomatic until 6–9 months of age when maternal IgG decreases. Present with recurrent infections with Streptococcus pneumoniae, Haemophilus influenzae, Mycoplasma pneumoniae, hepatitis virus, and enterovirus CNS infections. Examination shows lymphoid hypoplasia (tonsils and adenoids, no splenomegaly or lymphadenopathy). There is significant decrease in all immunoglobulins.
Genetics
Most antibodies are gamma globulins. Antibodies are made mainly by plasma cells, which are daughter cells of the B cell line. The Btk enzyme plays an essential role in the maturation of B cells in the bone marrow, and when mutated, immature pro-B lymphocytes are unable to develop into pre-B lymphocytes, which normally develop into mature (naive) B cells that leave the bone marrow into the blood stream.
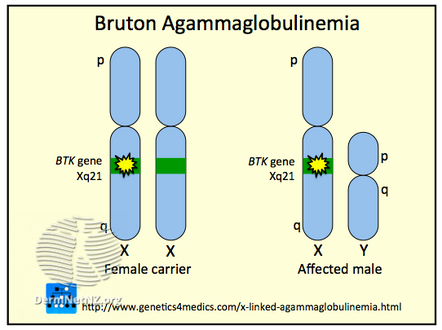
The disorder is inherited in an X-linked recessive fashion (as the gene linked to it is on the X chromosome) and is almost entirely limited to the sons of asymptomatic female carriers.[7] This is because males have only one copy of the X chromosome, while females have two copies; one normal copy of an X chromosome can compensate for mutations in the other X chromosome, so they are less likely to be symptomatic.
There is 30–50% chance of XLA patients having a positive family history of genetic inheritance. The rest of the cases occur as random mutations.[8] If a carrier female gives birth to a male child, there is a 50% chance that the male will have XLA. A carrier female has a 25% chance overall of giving birth to an affected male child. An XLA patient will pass on the gene, and all of his daughters will be XLA carriers, meaning that any male grandchildren from an XLA patient's daughters have a 50% chance of inheriting XLA. A female XLA patient can arise only as the child of an XLA patient and a carrier mother. XLA can also rarely result from a spontaneous mutation in the fetus of a non-carrier mother.
-
X-linked agammaglobulinemia
-

X-chr
.png)

Diagnosis
XLA diagnosis usually begins due to a history of recurrent infections, mostly in the respiratory tract, through childhood. This is due to humoral immunodeficiency.[8] The diagnosis is probable when blood tests show the complete lack of circulating B cells (determined by the B cell marker CD19 and/or CD20), as well as low levels of all antibody classes, including IgG, IgA, IgM, IgE and IgD.[7]
When XLA is suspected, it is possible to do a Western Blot test to determine whether the Btk protein is being expressed. Results of a genetic blood test confirm the diagnosis and will identify the specific Btk mutation,[7] however its cost prohibits its use in routine screening for all pregnancies. Women with an XLA patient in their family should seek genetic counseling before pregnancy. Although the symptoms of a XLA and other primary immune diseases (PID) include repeated and often severe infections, the average time for a diagnosis of a PID can be up to 10 years.[citation needed]
Treatment
The most common treatment for XLA is an intravenous infusion of immunoglobulin (IVIg, human IgG antibodies) every week, for life. IVIg is a human product extracted and pooled from thousands of blood donations. IVIg does not cure XLA but increases the patient's lifespan and quality of life, by generating passive immunity, and boosting the immune system.[7] With treatment, the number and severity of infections is reduced. With IVIg, XLA patients may live a relatively healthy life. A patient should attempt reaching a state where his IgG blood count exceeds 800 mg/kg. The dose is based on the patient's weight and IgG blood-count.
Muscle injections of immunoglobulin (IMIg) were common before IVIg was prevalent, but are less effective and much more painful; hence, IMIg is now uncommon. Subcutaneous treatment (SCIg) was recently approved by the U.S. Food and Drug Administration (FDA), which is recommended in cases of severe adverse reactions to the IVIg treatment.
Antibiotics are another common supplementary treatment. Local antibiotic treatment (drops, lotions) are preferred over systemic treatment (pills) for long-term treatment, if possible. One of the future prospects of XLA treatment is gene therapy, which could potentially cure XLA. Gene therapy technology is still in its infancy and may cause severe complications such as cancer and even death. Moreover, the long-term success and complications of this treatment are, as yet, unknown.
Other considerations
It is not recommended and dangerous for XLA patients to receive live attenuated vaccines such as live polio, or the measles, mumps, rubella (MMR vaccine).[7] Special emphasis is given to avoiding the oral live attenuated SABIN-type polio vaccine that has been reported to cause polio to XLA patients. Furthermore, it is not known if active vaccines in general have any beneficial effect on XLA patients as they lack normal ability to maintain immune memory.
XLA patients are specifically susceptible to viruses of the Enterovirus family, and mostly to: polio virus, coxsackie virus (hand, foot, and mouth disease) and Echoviruses. These may cause severe central nervous system conditions as chronic encephalitis, meningitis and death. An experimental anti-viral agent, pleconaril, is active against picornaviruses. XLA patients, however, are apparently immune to the Epstein-Barr virus (EBV), as they lack mature B cells (and so HLA co-receptors) needed for the viral infection.[9] Patients with XLA are also more likely to have a history of septic arthritis.[8]
It is not known if people with XLA are able to generate an allergic reaction, as they lack functional IgE antibodies. There is no special hazard for XLA patients in dealing with pets or outdoor activities.[7] Unlike in other primary immunodeficiencies XLA patients are at no greater risk for developing autoimmune illnesses.
Agammaglobulinemia (XLA) is similar to the primary immunodeficiency disorder hypogammaglobulinemia (CVID), and their clinical conditions and treatment are almost identical. However, while XLA is a congenital disorder, with known genetic causes, CVID may occur in adulthood and its causes are not yet understood. In addition, to X-linked agammaglobulinemia a couple of autosomal recessive agammaglobulinemia gene mutations have been described including mutations in IGHM [10], IGLL1[11], CD79A/B [12][13], BLNK [14] and deletion of the deletion of the terminal 14q32.33 chromosom.[15]
XLA was also historically mistaken as severe combined immunodeficiency (SCID), a much more severe immune deficiency ("Bubble boys").A strain of laboratory mouse, XID, is used to study XLA. These mice have a mutated version of the mouse Btk gene, and exhibit a similar, yet milder, immune deficiency as in XLA.[citation needed]
References
- ↑ 1.0 1.1 1.2 1.3 1.4 1.5 1.6 1.7 1.8 Lackey, AE; Ahmad, F (January 2020). "X-linked Agammaglobulinemia". PMID 31751055.
{{cite journal}}: Cite journal requires|journal=(help) - ↑ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. p. 83. ISBN 0-7216-2921-0.
- ↑ 3.00 3.01 3.02 3.03 3.04 3.05 3.06 3.07 3.08 3.09 3.10 "X-linked agammaglobulinemia | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 30 January 2021. Retrieved 28 January 2021.
- ↑ 4.0 4.1 4.2 4.3 4.4 4.5 4.6 4.7 "X-linked Agammaglobulinemia - Immunology; Allergic Disorders". Merck Manuals Professional Edition. Archived from the original on 30 July 2020. Retrieved 28 January 2021.
- ↑ Bruton OC (1952). "Agammaglobulinemia". Pediatrics. 9 (6): 722–8. PMID 14929630.. Reproduced in Buckley CR (1998). "Agammaglobulinemia, by Col. Ogden C. Bruton, MC, USA, Pediatrics, 1952;9:722-728". Pediatrics. 102 (1 Pt 2): 213–5. PMID 9651432.
- ↑ Mahdaviani, Seyed Alireza; Rezaei, Nima (2018). Pulmonary Manifestations of Primary Immunodeficiency Diseases. Springer. p. 78. ISBN 978-3-030-00880-2. Archived from the original on 2021-08-27. Retrieved 2021-01-28.
- ↑ 7.0 7.1 7.2 7.3 7.4 7.5 X-Linked Agammaglobulinemia Archived 2008-10-11 at the Wayback Machine Patient and Family Handbook for The Primary Immune Diseases. Third Edition. 2001. Published by the Immune Deficiency Foundation
- ↑ 8.0 8.1 8.2 Chun, Jin-Kyong; Lee, Taek Jin; Song, Jae Woo; Linton, John A; Kim, Dong Soo (2008-02-29). "Analysis of Clinical Presentations of Bruton Disease: A Review of 20 Years of Accumulated Data from Pediatric Patients at Severance Hospital". Yonsei Medical Journal. 49 (1): 28–36. doi:10.3349/ymj.2008.49.1.28. ISSN 0513-5796. PMC 2615253. PMID 18306466.
- ↑ Faulkner GC, Burrows SR, Khanna R, Moss DJ, Bird AG, Crawford DH (February 1999). "X-Linked agammaglobulinemia patients are not infected with Epstein-Barr virus: implications for the biology of the virus". Journal of Virology. 73 (2): 1555–64. doi:10.1128/JVI.73.2.1555-1564.1999. PMC 103980. PMID 9882361.
- ↑ "Archive copy". Archived from the original on 2020-05-11. Retrieved 2020-04-15.
{{cite web}}: CS1 maint: archived copy as title (link) - ↑ "Archive copy". Archived from the original on 2020-05-11. Retrieved 2020-04-15.
{{cite web}}: CS1 maint: archived copy as title (link) - ↑ "Archive copy". Archived from the original on 2020-05-11. Retrieved 2020-04-15.
{{cite web}}: CS1 maint: archived copy as title (link) - ↑ "Archive copy". Archived from the original on 2020-05-11. Retrieved 2020-04-15.
{{cite web}}: CS1 maint: archived copy as title (link) - ↑ "Archive copy". Archived from the original on 2020-05-11. Retrieved 2020-04-15.
{{cite web}}: CS1 maint: archived copy as title (link) - ↑ Geier, Christoph (October 2017). "Terminal 14q32.33 deletion as a novel cause of agammaglobulinemia". Clinical Immunology. Volume 183: Pages 41-45. PMID 28705765. Archived from the original on 2021-04-18. Retrieved 2020-04-15.
{{cite journal}}:|volume=has extra text (help)
External links
| Classification | |
|---|---|
| External resources |
- Pages with script errors
- CS1 errors: missing periodical
- Webarchive template wayback links
- CS1 maint: archived copy as title
- CS1 errors: extra text: volume
- CS1: long volume value
- All articles with unsourced statements
- Articles with unsourced statements from August 2016
- Articles with invalid date parameter in template
- Predominantly antibody deficiencies
- Neurocutaneous conditions
- X-linked recessive disorders
- RTT