Von Hippel–Lindau syndrome
| Von Hippel–Lindau syndrome | |
|---|---|
| Other names: Familial cerebello retinal angiomatosis[1] | |
 | |
| Typical distribution of hemangioblastomas in Von Hippel–Lindau disease | |
| Usual onset | 18-30 years[2] |
| Frequency | Rare,[3] males=females[2] |
Von Hippel–Lindau syndrome (VHL) is an inherited disease that presents with cysts and certain cancerous and benign tumors; typically of the nervous system, eyes, kidney, pancreas, endocrine system, and genital tract.[2] Symptoms depend on the size and location of tumor.[2] These tumors include hemangioblastomas of the central nervous system, causing headache, vomiting and unsteadiness.[2] If present in the eye, there may be visual loss.[2] There may be no symptoms in pheochromocytoma, or panic episodes, headaches, excess sweating and high blood pressure.[2] In inner ear endolymphatic sac tumors, the presentation might include ringing in the ears, hearing loss or vertigo.[2] Other tumors include kidney cancer, pancreatic serous cystadenoma and neuroendocrine tumors, paraganglioma, and genital tract papillary cystadenomas.[2] Average age of onset is in the 20s, though may occur between age 5-years and 65-years.[2]
It results from a mutation in the VHL gene on chromosome 3p25.3.[2] Diagnosis is by family history and either one or two of the associated tumors, laboratory tests and medical imaging.[2] Treatment and outcome depend on the type and size of tumor.[2]
VHL is rare.[3] In the United States it may occur in around 1 in 20,000 to 50,000 births.[2] It affects males and females equally.[2]
Signs and symptoms
Signs and symptoms associated with VHL disease include headaches, problems with balance and walking, dizziness, weakness of the limbs, vision problems, and high blood pressure. Conditions associated with VHL disease include angiomatosis, hemangioblastomas, pheochromocytoma, renal cell carcinoma, pancreatic cysts (pancreatic serous cystadenoma), endolymphatic sac tumor, and bilateral papillary cystadenomas of the epididymis (men) or broad ligament of the uterus (women).[4][5] Angiomatosis occurs in 37.2% of patients presenting with VHL disease and usually occurs in the retina. As a result, loss of vision is very common. However, other organs can be affected: strokes, heart attacks, and cardiovascular disease are common additional symptoms.[6] Approximately 40% of VHL disease presents with CNS hemangioblastomas and they are present in around 60-80%. Spinal hemangioblastomas are found in 13-59% of VHL disease and are specific because 80% are found in VHL disease.[7][8] Although all of these tumours are common in VHL disease, around half of cases present with only one tumour type.[8]
-
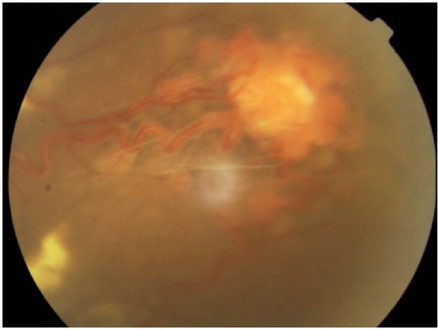
Fundus photograph of a retinal hemangioblastoma in a individual with Von Hippel-Lindau disease.
-

Slit lamp photograph showing retinal detachment in Von Hippel–Lindau disease


Pathogenesis
The disease is caused by mutations of the Von Hippel–Lindau tumor suppressor (VHL) gene on the short arm of chromosome 3 (3p25-26). There are over 1500 germline mutations and somatic mutations found in VHL disease.[9][10]

Every cell in the body has 2 copies of every gene (bar those found in the sex chromosomes, X and Y). In VHL disease, one copy of the VHL gene has a mutation and produces a faulty VHL protein (pVHL). However, the second copy still produces a functional protein. The condition is inherited in an autosomal dominant manner - one copy of the faulty gene is sufficient to increase the risk of developing tumours.[11][12]
Approximately 20% of cases of VHL disease are found in individuals without a family history, known as de novo mutations. An inherited mutation of the VHL gene is responsible for the remaining 80 percent of cases.[7]
30-40% of mutations in the VHL gene consist of 50-250kb deletion mutations that remove either part of the gene or the whole gene and flanking regions of DNA. The remaining 60-70% of VHL disease is caused by the truncation of pVHL by nonsense mutations, indel mutations or splice site mutations.[7]
VHL protein

The VHL protein (pVHL) is involved in the regulation of a protein known as hypoxia inducible factor 1α (HIF1α). This is a subunit of a heterodimeric transcription factor that at normal cellular oxygen levels is highly regulated. In normal physiological conditions, pVHL recognizes and binds to HIF1α only when oxygen is present due to the post translational hydroxylation of 2 proline residues within the HIF1α protein. pVHL is an E3 ligase that ubiquitinates HIF1α and causes its degradation by the proteasome. In low oxygen conditions or in cases of VHL disease where the VHL gene is mutated, pVHL does not bind to HIF1α. This allows the subunit to dimerise with HIF1β and activate the transcription of a number of genes, including vascular endothelial growth factor, platelet-derived growth factor B, erythropoietin and genes involved in glucose uptake and metabolism.[12][13] A new novel missense mutation in VHL genes c.194 C>T, c.239 G>A, c.278 G>A, c.319 C>G, c.337 C>G leading to the following variations p.Ala 65 Val, p.Gly 80 Asp, p.Gly 93 Glu, p.Gln 107 Glu, p.Gln 113 Glu[check spelling] in the protein contributed to renal clear cell carcinoma.[14]
Diagnosis
The detection of tumours specific to VHL disease is important in the disease's diagnosis. In individuals with a family history of VHL disease, one hemangioblastoma, pheochromocytoma or renal cell carcinoma may be sufficient to make a diagnosis. As all the tumours associated with VHL disease can be found sporadically, at least two tumours must be identified to diagnose VHL disease in a person without a family history.[7][8]
Genetic diagnosis is also useful in VHL disease diagnosis. In hereditary VHL disease, techniques such as the Southern blot and gene sequencing can be used to analyse DNA and identify mutations. These tests can be used to screen family members of those afflicted with VHL disease; de novo cases that produce genetic mosaicism are more difficult to detect because mutations are not found in the white blood cells that are used for genetic analysis.[7][15]
Classification
VHL disease can be subdivided according to the clinical manifestations, although these groups often correlate with certain types of mutations present in the VHL gene.[16]
Treatment
Early recognition and treatment of specific manifestations of VHL can substantially decrease complications and improve quality of life. For this reason, individuals with VHL disease are usually screened routinely for retinal angiomas, CNS hemangioblastomas, clear-cell renal carcinomas and pheochromocytomas.[17] CNS hemangioblastomas are usually surgically removed if they are symptomatic. Photocoagulation and cryotherapy are usually used for the treatment of symptomatic retinal angiomas, although anti-angiogenic treatments may also be an option. Renal tumours may be removed by a partial nephrectomy or other techniques such as radiofrequency ablation.[7]
Belzutifan is a drug under investigation for the treatment of von Hippel–Lindau disease-associated renal cell carcinoma.[18][19][20][21]
Epidemiology
VHL disease has an incidence of one in 36,000 births. There is over 90% penetrance by the age of 65.[22] Age at diagnosis varies from infancy to age 60–70 years, with an average patient age at clinical diagnosis of 26 years.[citation needed]
History

The German ophthalmologist Eugen von Hippel first described angiomas in the eye in 1904.[23] Arvid Lindau described the angiomas of the cerebellum and spine in 1927.[24] The term Von Hippel–Lindau disease was first used in 1936; however, its use became common only in the 1970s.[7]
Notable cases
Some descendants of the McCoy family (involved in the Hatfield-McCoy feud of Appalachia, USA) are presumed to have VHL. In an article appearing in the Associated Press, it has been speculated by a Vanderbilt University endocrinologist that the hostility underlying the Hatfield–McCoy feud may have been partly due to the consequences of Von Hippel–Lindau disease. The article suggests that the McCoy family was predisposed to bad tempers because many of them had a pheochromocytoma which produced excess adrenaline and a tendency toward explosive tempers.[25]
Nomenclature
Other uncommon names are: angiomatosis retinae, familial cerebello-retinal angiomatosis, cerebelloretinal hemangioblastomatosis, Hippel Disease, Hippel–Lindau syndrome, HLS, VHL, Lindau disease or retinocerebellar angiomatosis.[26][27]
See also
References
- ↑ RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Von Hippel Lindau disease". www.orpha.net. Archived from the original on 8 February 2019. Retrieved 25 May 2019.
- ↑ 2.00 2.01 2.02 2.03 2.04 2.05 2.06 2.07 2.08 2.09 2.10 2.11 2.12 2.13 2.14 WHO Classification of Tumours Editorial Board, ed. (2020). "16. Genetic tumour syndromes of the female genital tract". Female genital tumours: WHO Classification of Tumours. Vol. 4 (5th ed.). Lyon (France): International Agency for Research on Cancer. p. 560. ISBN 978-92-832-4504-9. Archived from the original on 2022-06-17. Retrieved 2023-10-21.
- ↑ 3.0 3.1 "Von Hippel-Lindau disease | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 2018-04-18. Retrieved 2018-04-17.
- ↑ Lindsay, Kenneth W; Ian Bone; Robin Callander; J. van Gijn (1991). Neurology and Neurosurgery Illustrated. United States: Churchill Livingstone. ISBN 978-0-443-04345-1.
- ↑ Frantzen, Carlijn; Links, Thera P.; Giles, Rachel H. (21 June 2012). "Von Hippel-Lindau Syndrome". Von Hippel-Lindau Disease. GeneReviews at NCBI. University of Washington, Seattle. Archived from the original on 23 February 2017. Retrieved 30 March 2013.
- ↑ Wong WT; Agrón E; Coleman HR; et al. (February 2007). "Genotype–phenotype correlation in von Hippel–Lindau disease with retinal angiomatosis". Archives of Ophthalmology. 125 (2): 239–45. doi:10.1001/archopht.125.2.239. PMC 3019103. PMID 17296901. Archived from the original on 2008-12-12. Retrieved 2008-10-22.
- ↑ 7.0 7.1 7.2 7.3 7.4 7.5 7.6 Maher ER; Glenn GM; Walther M; et al. (June 2011). "von Hippel-Lindau disease: a clinical and scientific review". European Journal of Human Genetics. 19 (6): 617–23. doi:10.1038/ejhg.2010.175. PMC 3110036. PMID 21386872.
- ↑ 8.0 8.1 8.2 Friedrich, CA (December 1, 1999). "Von Hippel-Lindau syndrome. A pleomorphic condition". Cancer. 86 (11 Suppl): 2478–82. doi:10.1002/(SICI)1097-0142(19991201)86:11+<2478::AID-CNCR4>3.0.CO;2-5. PMID 10630173.
- ↑ Kondo, K; Kaelin Jr, WG (10 March 2001). "The von Hippel–Lindau Tumor Suppressor Gene". Experimental Cell Research. 264 (1): 117–125. doi:10.1006/excr.2000.5139. PMID 11237528.
- ↑ Nordstrom-O'Brien M; van der Luijt RB; van Rooijen E; et al. (May 2010). "Genetic analysis of von Hippel-Lindau disease". Hum. Mutat. 31 (5): 521–37. doi:10.1002/humu.21219. PMID 20151405. S2CID 38910112.
- ↑ Knudson, AG (Nov 2001). "Two genetic hits (more or less) to cancer". Nature Reviews Cancer. 1 (2): 157–62. doi:10.1038/35101031. PMID 11905807. S2CID 20201610.
- ↑ 12.0 12.1 Kaelin, WG (2007). "Von Hippel-Lindau disease". Annual Review of Pathology. 2: 145–73. doi:10.1146/annurev.pathol.2.010506.092049. PMID 18039096.
- ↑ Bader, HL; Hsu, T (June 4, 2012). "Systemic VHL gene functions and the VHL disease". FEBS Letters. 586 (11): 1562–9. doi:10.1016/j.febslet.2012.04.032. PMC 3372859. PMID 22673568.
- ↑ Kumar, P. S.; Venkatesh, K.; Srikanth, L.; Sarma, P. V.; Reddy, A. R.; Subramanian, S.; Phaneendra, B. V. (July 2013). "Novel three missense mutations observed in Von Hippel-Lindau gene in a patient reported with renal cell carcinoma". Indian Journal of Human Genetics. 19 (3): 373–376. doi:10.4103/0971-6866.120809. PMC 3841571. PMID 24339559.
- ↑ Lonser RR (June 2003). "von Hippel-Lindau disease". Lancet. 361 (9374): 2059–67. doi:10.1016/S0140-6736(03)13643-4. PMID 12814730. S2CID 13783714.
- ↑ Calzada, MJ (March 2010). "Von Hippel-Lindau syndrome: molecular mechanisms of the disease". Clinical & Translational Oncology. 12 (3): 160–5. doi:10.1007/s12094-010-0485-9. PMID 20231120. S2CID 7789108.
- ↑ Priesemann M; Davies KM; Perry LA; et al. (2006). "Benefits of screening in von Hippel-Lindau disease – comparison of morbidity associated with initial tumours in affected parents and children". Horm. Res. 66 (1): 1–5. doi:10.1159/000093008. PMID 16651847. S2CID 29862078.
- ↑ "Belzutifan". SPS - Specialist Pharmacy Service. 18 March 2021. Archived from the original on 26 April 2021. Retrieved 25 April 2021.
- ↑ "MHRA awards first 'innovation passport' under new pathway". RAPS (Press release). Archived from the original on 26 April 2021. Retrieved 25 April 2021.
- ↑ "Merck Receives Priority Review From FDA for New Drug Application for HIF-2α Inhibitor Belzutifan (MK-6482)" (Press release). Merck. 16 March 2016. Archived from the original on 18 March 2021. Retrieved 25 April 2021 – via Business Wire.
- ↑ "FDA Grants Priority Review to Belzutifan for von Hippel-Lindau Disease–Associated RCC". Cancer Network. Archived from the original on 2021-04-26. Retrieved 2021-04-26.
- ↑ Kim, JJ; Rini, BI; Hansel, DE (2010). Von Hippel Lindau syndrome. Advances in Experimental Medicine and Biology. Vol. 685. pp. 228–49. doi:10.1007/978-1-4419-6448-9_22. ISBN 978-1-4419-6447-2. PMID 20687511.
- ↑ Von Hippel E (1904). "Ueber eine sehr seltene Erkrankung der Netzhaut". Albrecht von Graefes Arch Ophthal. 59: 83–106. doi:10.1007/bf01994821. S2CID 22425158. Archived from the original on 2022-01-04. Retrieved 2022-04-04.
- ↑ Lindau A (1927). "Zur Frage der Angiomatosis Retinae und Ihrer Hirncomplikation". Acta Ophthalmol. 4 (1–2): 193–226. doi:10.1111/j.1755-3768.1926.tb07786.x. S2CID 73385451.
- ↑ "Hatfield–McCoy feud blamed on 'rage' disease". MSNBC.com. 2007-04-05. Archived from the original on 2007-04-07. Retrieved 2007-04-05.
- ↑ "Von Hippel-Lindau Disease". Rare Disease Database. NORD - National Organization for Rare Disorders. 2019. Archived from the original on 25 April 2015. Retrieved 4 April 2022.
- ↑ "von Hippel-Lindau Disease". Medical Subject Headings (MeSH). U.S. National Library of Medicine. Archived from the original on 6 December 2021. Retrieved 4 April 2022.
External links
| Classification | |
|---|---|
| External resources |
- GeneReviews/NCBI/NIH/UW entry on Von Hippel-Lindau Syndrome Archived 2017-02-23 at the Wayback Machine
- Von Hippel–Lindau Disease (VHL) at NINDS
- Von Hippel–Lindau syndrome at NLM Genetics Home Reference
- von Hippel–Lindau syndrome at CHORUS
- Hippel–Lindau disease at Who Named It?
- Online Mendelian Inheritance in Man (OMIM): 608537 (VHL gene)
- Pages with script errors
- All articles with unidentified words
- Articles with unidentified words from May 2019
- Articles with invalid date parameter in template
- All articles with unsourced statements
- Articles with unsourced statements from May 2019
- Webarchive template wayback links
- Autosomal dominant disorders
- Rare diseases
- Hereditary cancers
- Genodermatoses
- Syndromes