DiGeorge syndrome
| DiGeorge syndrome | |
|---|---|
| Other names: DiGeorge anomaly,[1][2] velocardiofacial syndrome (VCFS),[3] Shprintzen syndrome,[4] conotruncal anomaly face syndrome (CTAF),[5] Takao syndrome,[6] Sedlackova syndrome,[7] Cayler cardiofacial syndrome,[7] CATCH22,[7] 22q11.2 deletion syndrome[7] | |
 | |
| A child with characteristic facial features of DiGeorge syndrome | |
| Specialty | Medical genetics |
| Symptoms | Varied; commonly congenital heart problems, specific facial features, cleft palate[7] |
| Complications | Kidney problems, hearing loss, autoimmune disorders[7] |
| Causes | Genetic (typically new mutation)[7] |
| Diagnostic method | Based on symptoms and genetic testing[5] |
| Differential diagnosis | Smith–Lemli–Opitz syndrome, Alagille syndrome, VACTERL, Oculo-auriculo-vertebral spectrum[5] |
| Treatment | Involves many healthcare specialties[5] |
| Prognosis | Depends on the specific symptoms[3] |
| Frequency | 1 in 4,000[7] |
DiGeorge syndrome, also known as 22q11.2 deletion syndrome, is a syndrome caused by the deletion of a small segment of chromosome 22.[7] While the symptoms can vary, they often include congenital heart problems, specific facial features, frequent infections, developmental delay, learning problems and cleft palate.[7] Associated conditions include kidney problems, hearing loss and autoimmune disorders such as rheumatoid arthritis or Graves' disease.[7]
DiGeorge syndrome is typically due to the deletion of 30 to 40 genes in the middle of chromosome 22 at a location known as 22q11.2.[3] About 90% of cases occur due to a new mutation during early development, while 10% are inherited from a person's parents.[7] It is autosomal dominant, meaning that only one affected chromosome is needed for the condition to occur.[7] Diagnosis is suspected based on the symptoms and confirmed by genetic testing.[5]
Although there is no cure, treatment can improve symptoms.[3] This often includes a multidisciplinary approach with efforts to improve the function of the potentially many organ systems involved.[8] Long-term outcomes depend on the symptoms present and the severity of the heart and immune system problems.[3] With treatment, life expectancy may be normal.[9]
DiGeorge syndrome occurs in about 1 in 4,000 people.[7] The syndrome was first described in 1968 by American physician Angelo DiGeorge.[10][11] In late 1981, the underlying genetics were determined.[11]
Signs and symptoms

The features of this syndrome vary widely, even among members of the same family, and affect many parts of the body. Characteristic signs and symptoms may include birth defects such as congenital heart disease, defects in the palate, most commonly related to neuromuscular problems with closure (velopharyngeal insufficiency), learning disabilities, mild differences in facial features, and recurrent infections. Infections are common in children due to problems with the immune system's T cell-mediated response that in some patients is due to an absent or hypoplastic thymus. DiGeorge syndrome may be first spotted when an affected newborn has heart defects or convulsions from hypocalcemia due to malfunctioning parathyroid glands and low levels of parathyroid hormone (parathormone).
Affected individuals may also have other kinds of birth defects including kidney abnormalities and significant feeding difficulties as babies. Gastrointestinal issues are also very common in this patient population. Digestive motility issues may result in constipation.[12] Disorders such as hypothyroidism and hypoparathyroidism or thrombocytopenia (low platelet levels), and psychiatric illnesses are common late-occurring features.[13]
Microdeletions in chromosomal region 22q11.2 are associated with a 20 to 30-fold increased risk of schizophrenia.[14] Studies provide various rates of 22q11.2DS in schizophrenia, ranging from 0.5 to 2.0% and averaging about 1.0%, compared with the overall estimated 0.025% risk of the 22q11.2DS in the general population.[15]
Salient features can be summarized using the mnemonic CATCH-22 to describe 22q11.2DS, with the 22 signifying the chromosomal abnormality is found on the 22nd chromosome, as below:[16]
- Cardiac abnormality (commonly interrupted aortic arch, truncus arteriosus and tetralogy of Fallot)
- Abnormal facies
- Thymic aplasia
- Cleft palate
- Hypocalcemia/hypoparathyroidism
Individuals can have many possible features, ranging in number of associated features and from the mild to the very serious. Symptoms shown to be common include:
- Congenital heart disease (40% of individuals), particularly conotruncal malformations (interrupted aortic arch (50%), persistent truncus arteriosus (34%), tetralogy of Fallot, and ventricular septal defect)
- Cyanosis (bluish skin due to poor circulation of oxygen-rich blood)
- Palatal abnormalities (50%), particularly velopharyngeal incompetence, submucosal cleft palate, and cleft palate; characteristic facial features (present in the majority of Caucasian individuals) including hypertelorism
- Learning difficulties (90%), including cognitive deficits, attention deficit disorders[17]
- Hypocalcemia (50%)(due to hypoparathyroidism)
- Significant feeding problems (30%)
- Kidney anomalies (37%)
- Hearing loss (both conductive and sensorineural) (hearing loss with craniofacial syndromes)
- Laryngotracheoesophageal anomalies
- Growth hormone deficiency
- Autoimmune disorders
- Immune disorders due to reduced T cell numbers
- Seizures (with or without hypocalcemia)
- Skeletal abnormalities
- Psychiatric disorders[17]
This syndrome is characterized by incomplete penetrance. Therefore, there is a marked variability in clinical expression between the different patients. This often makes early diagnosis difficult.[18]
Cognitive impairments
Children with DiGeorge syndrome have a specific profile in neuropsychological tests. They usually have a below-borderline normal IQ, with most individuals having higher scores in the verbal than the nonverbal domains. Some are able to attend normal schools, while others are home-schooled or in special classes. The severity of hypocalcemia early in childhood is associated with autism-like behavioral difficulties.[19]
Adults with DiGeorge syndrome are a specifically high-risk group for developing schizophrenia. About 30% have at least one incident of psychosis and about a quarter develop actual schizophrenia.[20]
Individuals with DiGeorge syndrome also have a higher risk of developing early onset Parkinson's disease (PD). Diagnosis of Parkinson's can be delayed by up to 10 years due to the use of antipsychotics, which can cause parkinsonian symptoms.[21][22]
Speech and language
Current research demonstrates a unique profile of speech and language impairments is associated with 22q11.2DS. Children often perform lower on speech and language evaluations in comparison to their nonverbal IQ scores.[contradictory] Common problems include hypernasality, language delays, and speech sound errors.[23][24][25]
Hypernasality occurs when air escapes through the nose during the production of oral speech sounds, resulting in reduced intelligibility. This is a common characteristic in the speech and language profile because 69% of children have palatal abnormalities. If the structure of the soft palate velum is such that it does not stop the flow of air from going up to the nasal cavity, it will cause hypernasal speech. This phenomenon is referred as velopharyngeal inadequacy (VPI). Hearing loss can also contribute to increased hypernasality because children with hearing impairments can have difficulty self monitoring their oral speech output. The treatment options available for VPI include prosthesis and surgery.[23][24][26][27][28]
Difficulties acquiring vocabulary and formulating spoken language (expressive language deficits) at the onset of language development are also part of the speech and language profile associated with the 22q11.2 deletion. Vocabulary acquisition is often severely delayed for preschool-age children. In some recent studies, children had a severely limited vocabulary or were still not verbal at 2–3 years of age. School-age children do make progress with expressive language as they mature, but many continue to have delays and demonstrate difficulty when presented with language tasks such as verbally recalling narratives and producing longer and more complex sentences. Receptive language, which is the ability to comprehend, retain, or process spoken language, can also be impaired, although not usually with the same severity as expressive language impairments.[24][27][28][29]
Articulation errors are commonly present in children with DiGeorge syndrome. These errors include a limited phonemic (speech sound) inventory and the use of compensatory articulation strategies resulting in reduced intelligibility. The phonemic inventory typically produced consists of sounds made in the front or back of the oral cavity such as: /p/, /w/, /m/, /n/, and glottal stops. Sound made in the middle of the mouth are completely absent. Compensatory articulation errors made by this population of children include: glottal stops, nasal substitutions, pharyngeal fricatives, linguapalatal sibilants, reduced pressure on consonant sounds, or a combination of these symptoms. Of these errors, glottal stops have the highest frequency of occurrence. It is reasoned that a limited phonemic inventory and the use of compensatory articulation strategies is present due to the structural abnormalities of the palate. The speech impairments exhibited by this population are more severe during the younger ages and show a trend of gradual improvement as the child matures.[23][27]
Genetics

DiGeorge syndrome is caused by a heterozygous deletion of part of the long arm (q) of chromosome 22, region 1, band 1, sub-band 2 (22q11.2). Approximately 80-90% of patients have a deletion of 3 Mb and 8% have a deletion of 1.5Mb.[30][31] The number of genes affected by the deletion has been cited as approximately 30 to 50.[32][33] Very rarely, patients with somewhat similar clinical features may have deletions on the short arm of chromosome 10.[34] The disorder has an autosomal dominant inheritance pattern.
A French study of 749 people diagnosed between 1995 and 2013 found that the mutation was inherited in 15% of patients, of which 85.5% was from the mother.[35] Other studies have found inheritance rates of 6-10%. The majority cases are a result of a de novo (new to the family) deletion.[12] This is because the 22q11 region has a structure that makes it highly prone to rearrangements during sperm or egg formation.[36]
The exact mechanism that causes all of the associated features of the syndrome is unknown.[30] Of the 30–50 genes in the deleted region, a number have been identified as possibly playing a role in the development of some of the signs and symptoms.
TBX1
Haploinsufficiency of the TBX1 gene (T-box transcription factor TBX1) is thought to be the cause of some of the symptoms observed. Point mutations in this gene have also been observed in individuals with DiGeorge syndrome.[30] TBX1 is part of the T-box family of genes which have an important role in tissue and organ formation during embryonic development and it may have a role in the regulation of differentiation of post migration neural crest cells. The neural crest forms many of the structures affected in DiGeorge syndrome, including the skull bones, mesenchyme of the face and palate, the outflow tract of the heart, and the thymus and parathyroid stroma. When there is a loss of expression of FGF18 during the development of the pharyngeal arches, neural crest cell death is seen. Although neither FGF18 or TBX1 are expressed in the neural crest cells, TBX1 might have a role in the regulation of FGF18 expression, ensuring that the differentiation of these cells in the pharyngeal region is correct. Therefore, dysfunction of TBX1 may be responsible for some of the symptoms in DiGeorge syndrome.[31]
Research in mouse models has shown that deletion of Tbx1 leads to several defects similar to those seen in humans, mainly affecting development of the great arteries and the thymus.[37][38]
The abnormalities seen in the great arteries of mice deficient of Tbx1 are a consequence of abnormal formation and remodelling of the aortic arches during early development. The role of Tbx1 for correct formation and remodelling of the aortic arches has been extensively studied in various mouse models suggesting the key role of Tbx1 for cardiovascular development and the phenotypes seen in DiGeorge syndrome.
DGCR8
In mice, haploinsufficiency of the DGCR8 gene has been linked to improper regulation of the microRNA miR-338 and 22q11.2 deletion phenotypes. [39]
TANGO2
Transport and golgi organization 2 homolog (TANGO2) also known as chromosome 22 open reading frame 25 (C22orf25) is a protein that in humans is encoded by the TANGO2 gene.
The gene coding for C22orf25 is located on chromosome 22 and the location q11.21, so it is often associated with 22q11.2 deletion syndrome.[40] But with TANGO2 disorder being autosomal recessive, will not occur in all cases.
Mutations in the TANGO2 gene may cause defects in mitochondrial β-oxidation[41] and increased endoplasmic reticulum stress and a reduction in Golgi volume density.[42] These mutations results in early onset hypoglycemia, hyperammonemia, rhabdomyolysis, cardiac arrhythmias, and encephalopathy that later develops into cognitive impairment.[41][42]
Parkinson's disease genes
22q11.2DS has been associated with a higher risk of early onset Parkinson's disease (PD). The neuropathology seen is similar to LRRK2-associated PD. None of the genes affected in individuals with 22q11.2DS have previously been linked to PD but there are a number that are likely candidates. These include DGCR8 which is important for biogenesis of brain microDNA, SRPT5 which encodes a protein that interacts with the PARK2 protein, COMT which is involved in regulating dopamine levels, and microRNA miR-185 which is thought to target known PD loci LRRK2.[21]
Diagnosis
Diagnosis of DiGeorge syndrome can be difficult due to the number of potential symptoms and the variation in phenotypes between individuals. It is suspected in patients with one or more signs of the deletion. In these cases a diagnosis of 22q11.2DS is confirmed by observation of a deletion of part of the long arm (q) of chromosome 22, region 1, band 1, sub-band 2. Genetic analysis is normally performed using fluorescence in situ hybridization (FISH), which is able to detect microdeletions that standard karyotyping (e.g. G-banding) miss. Newer methods of analysis include Multiplex ligation-dependent probe amplification assay (MLPA) and quantitative polymerase chain reaction (qPCR), both of which can detect atypical deletions in 22q11.2 that are not detected by FISH.[43] qPCR analysis is also quicker than FISH, which can have a turn around of 3 to 14 days.[12]
A 2008 study of a new high-definition MLPA probe developed to detect copy number variation at 37 points on chromosome 22q found it to be as reliable as FISH in detecting normal 22q11.2 deletions. It was also able to detect smaller atypical deletions that are easily missed using FISH. These factors, along with the lower expense and easier testing mean that this MLPA probe could replace FISH in clinical testing.[44]
Genetic testing using BACs-on-Beads has been successful in detecting deletions consistent with 22q11.2DS during prenatal testing.[45][46] Array-comparative genomic hybridization (array-CGH) uses a large number of probes embossed in a chip to screen the entire genome for deletions or duplications. It can be used in post and pre-natal diagnosis of 22q11.2.[47]
Fewer than 5% of individuals with symptoms of DiGeorge syndrome have normal routine cytogenetic studies and negative FISH testing. In these cases, atypical deletions are the cause.[48] Some cases of 22q11.2 deletion syndrome have defects in other chromosomes, notably a deletion in chromosome region 10p14.[34]
-
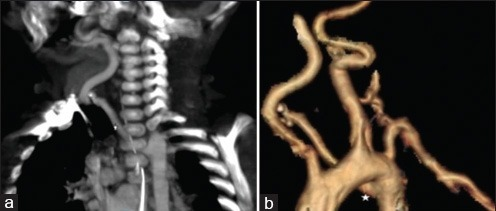
CT shows aberrant right subclavian artery
-

Result of FISH analysis using LSI probe (TUPLE 1) from DiGeorge/velocardiofacial syndrome critical region. TUPLE 1 (HIRA) probe was labeled in Spectrum Orange and Arylsulfatase A (ARSA) in Spectrum Green as control. Absence of the orange signal indicates deletion of the TUPLE 1 locus at 22q11.2.
-
![Brain CT in DiGeorge syndrome, demonstrating basal ganglia and periventricular calcification.[49]](https://upload.wikimedia.org/wikipedia/commons/thumb/e/ec/Brain_computer_tomography_cuts_of_the_patient_with_22q11.2_syndrome%2C_demonstrating_basal_ganglia_and_periventricular_calcification.jpg/434px-Brain_computer_tomography_cuts_of_the_patient_with_22q11.2_syndrome%2C_demonstrating_basal_ganglia_and_periventricular_calcification.jpg)
Brain CT in DiGeorge syndrome, demonstrating basal ganglia and periventricular calcification.[49]


![Brain CT in DiGeorge syndrome, demonstrating basal ganglia and periventricular calcification.[49]](/wiki/File:Brain_computer_tomography_cuts_of_the_patient_with_22q11.2_syndrome,_demonstrating_basal_ganglia_and_periventricular_calcification.jpg)
Treatment
No cure is known for DiGeorge syndrome. Certain individual features are treatable using standard treatments. The key is to identify each of the associated features and manage each using the best available treatments.
For example, in children, it is important that the immune problems are identified early, as special precautions are required regarding blood transfusion and immunization with live vaccines.[50] Thymus transplantation can be used to address absence of the thymus in the rare, so-called "complete" DiGeorge syndrome.[51] Bacterial infections are treated with antibiotics. Cardiac surgery is often required for congenital heart abnormalities. Hypoparathyroidism causing hypocalcaemia often requires lifelong vitamin D and calcium supplements. Specialty clinics that provide multi-system care allow for individuals with DiGeorge syndrome to be evaluated for all of their health needs and allow for careful monitoring of the patients. An example of this type of system is the 22q Deletion Clinic at SickKids Hospital in Toronto, Canada, which provides children with 22q11 deletion syndrome ongoing support, medical care and information from a team of health care workers.[52]
Epidemiology
DiGeorge syndrome is estimated to affect between one in 2000 and one in 4000 live births.[53][54] This estimate is based on major birth defects and may be an underestimate, because some individuals with the deletion have few symptoms and may not have been formally diagnosed. It is one of the most common causes of intellectual disability due to a genetic deletion syndrome.[55]
The number of people affected has been expected to rise because of multiple reasons: (1) surgical and medical advances, an increasing number of people are surviving heart defects associated with the syndrome. These individuals are in turn having children. The chances of a person with DiGeorge syndrome having an affected child is 50% for each pregnancy; (2) Parents who have affected children, but who were unaware of their own genetic conditions, are now being diagnosed as genetic testing become available; (3) Molecular genetics techniques such as FISH (fluorescence in situ hybridization) have limitations and have not been able to detect all 22q11.2 deletions. Newer technologies have been able to detect these atypical deletions.[56]
Name
The signs and symptoms of DiGeorge syndrome are so varied that different groupings of its features were once regarded as separate conditions. These original classifications included velocardiofacial syndrome, Shprintzen syndrome, DiGeorge sequence/syndrome, Sedlackova syndrome, and conotruncal anomaly face syndrome. All are now understood to be presentations of a single syndrome.
ICD-10 2015 version mentions DiGeorge syndrome using two codes: D82.1 (Di George syndrome)[57] and Q93.81 (Velo-cardio-facial syndrome).[58] The ICD-11 Beta Draft discusses the syndrome under “LD50.P1 CATCH 22 phenotype".[58] However, since this syndrome is caused by the deletion of a small piece of chromosome 22, some recommend that the name "22q11.2 deletion syndrome (22q11.2DS)" be used.[59][12] Some experts support changing the name of both DiGeorge and velocardiofacial syndromes to CATCH-22.[citation needed] The International 22q11.2 Foundation, through its "Same Name Campaign", advocates for the name 22q11.2 deletion syndrome.[60]
See also
References
- ↑ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 978-1-4160-2999-1.
- ↑ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 978-0-7216-2921-6.
- ↑ 3.0 3.1 3.2 3.3 3.4 "22q11.2 deletion syndrome". Genetic and Rare Diseases Information Center (GARD). Archived from the original on 5 July 2017. Retrieved 15 May 2017.
- ↑ Shprintzen RJ, Goldberg RB, Lewin ML, Sidoti EJ, Berkman MD, Argamaso RV, Young D (January 1978). "A new syndrome involving cleft palate, cardiac anomalies, typical facies, and learning disabilities: velo-cardio-facial syndrome". Cleft Palate J. 15 (1): 56–62. PMID 272242.
- ↑ 5.0 5.1 5.2 5.3 5.4 "Chromosome 22q11.2 Deletion Syndrome - NORD (National Organization for Rare Disorders)". NORD (National Organization for Rare Disorders). 2017. Archived from the original on 28 January 2017. Retrieved 10 July 2017.
- ↑ Burn J, Takao A, Wilson D, Cross I, Momma K, Wadey R, Scambler P, Goodship J (October 1993). "Conotruncal anomaly face syndrome is associated with a deletion within chromosome 22q11". J. Med. Genet. 30 (10): 822–4. doi:10.1136/jmg.30.10.822. PMC 1016562. PMID 8230157.
- ↑ 7.00 7.01 7.02 7.03 7.04 7.05 7.06 7.07 7.08 7.09 7.10 7.11 7.12 7.13 "22q11.2 deletion syndrome". Genetics Home Reference. July 2013. Archived from the original on 13 May 2017. Retrieved 15 May 2017.
- ↑ Kobrynski LJ, Sullivan KE (October 2007). "Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes". Lancet. 370 (9596): 1443–52. doi:10.1016/S0140-6736(07)61601-8. PMID 17950858.
- ↑ Goldman, Lee; Schafer, Andrew I. (2015). Goldman-Cecil Medicine E-Book. Elsevier Health Sciences. p. 702. ISBN 9780323322850. Archived from the original on 2017-11-05.
- ↑ DiGeorge, A (1968). "Congenital absence of the thymus and its immunologic consequences: concurrence with congenital hypoparathyroidism". March of Dimes-Birth Defects Foundation: 116–21.
- ↑ 11.0 11.1 Restivo A, Sarkozy A, Digilio MC, Dallapiccola B, Marino B (February 2006). "22q11 deletion syndrome: a review of some developmental biology aspects of the cardiovascular system". J Cardiovasc Med (Hagerstown). 7 (2): 77–85. doi:10.2459/01.JCM.0000203848.90267.3e. PMID 16645366.
- ↑ 12.0 12.1 12.2 12.3 McDonald-McGinn DM, Sullivan KE (January 2011). "Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome)". Medicine (Baltimore). 90 (1): 1–18. doi:10.1097/MD.0b013e3182060469. PMID 21200182.
- ↑ Debbané M, Glaser B, David MK, Feinstein C, Eliez S (2006). "Psychotic symptoms in children and adolescents with 22q11.2 deletion syndrome: Neuropsychological and behavioral implications". Schizophr. Res. 84 (2–3): 187–93. doi:10.1016/j.schres.2006.01.019. PMID 16545541.
- ↑ [non-primary source needed] Bassett AS, Chow EW, AbdelMalik P, Gheorghiu M, Husted J, Weksberg R (2003). "The schizophrenia phenotype in 22q11 deletion syndrome". Am J Psychiatry. 160 (9): 1580–6. doi:10.1176/appi.ajp.160.9.1580. PMC 3276594. PMID 12944331.
- ↑ [non-primary source needed] Horowitz A, Shifman S, Rivlin N, Pisanté A, Darvasi A (2005). "A survey of the 22q11 microdeletion in a large cohort of schizophrenia patients". Schizophr. Res. 73 (2–3): 263–7. doi:10.1016/j.schres.2004.02.008. PMID 15653270.
- ↑ Burn J (October 1999). "Closing time for CATCH22". J. Med. Genet. 36 (10): 737–8. doi:10.1136/jmg.36.10.737. PMC 1734243. PMID 10528851. Archived from the original on 2021-04-22. Retrieved 2011-10-25.
- ↑ 17.0 17.1 Lindsay EA (November 2001). "Chromosomal microdeletions: dissecting del22q11 syndrome". Nat. Rev. Genet. 2 (11): 858–68. doi:10.1038/35098574. PMID 11715041.
- ↑ Swillen A, Vogels A, Devriendt K, Fryns JP (2000). "Chromosome 22q11 deletion syndrome: update and review of the clinical features, cognitive-behavioral spectrum, and psychiatric complications". Am. J. Med. Genet. 97 (2): 128–35. doi:10.1002/1096-8628(200022)97:2<128::AID-AJMG4>3.0.CO;2-Z. PMID 11180220.
- ↑ Muldoon M, Ousley OY, Kobrynski LJ, Patel S, Oster ME, Fernandez-Carriba S, Cubells JF, Coleman K, Pearce BD (September 2015). "The effect of hypocalcemia in early childhood on autism-related social and communication skills in patients with 22q11 deletion syndrome". Eur Arch Psychiatry Clin Neurosci. 265 (6): 519–24. doi:10.1007/s00406-014-0546-0. PMC 4379129. PMID 25267002.
- ↑ Zinkstok J, van Amelsvoort T (2005). "Neuropsychological profile and neuroimaging in patients with 22Q11.2 Deletion Syndrome: a review". Child Neuropsychol. 11 (1): 21–37. doi:10.1080/09297040590911194. PMID 15823981.
- ↑ 21.0 21.1 Butcher NJ, Kiehl TR, Hazrati LN, Chow EW, Rogaeva E, Lang AE, Bassett AS (2013). "Association between early-onset Parkinson disease and 22q11.2 deletion syndrome: identification of a novel genetic form of Parkinson disease and its clinical implications". JAMA Neurol. 70 (11): 1359–66. doi:10.1001/jamaneurol.2013.3646. PMC 4464823. PMID 24018986.
- ↑ Mok KY, Sheerin U, Simón-Sánchez J, Salaka A, Chester L, Escott-Price V, et al. (May 2016). "Deletions at 22q11.2 in idiopathic Parkinson's disease: a combined analysis of genome-wide association data". Lancet Neurol. 15 (6): 585–96. doi:10.1016/S1474-4422(16)00071-5. PMC 4828586. PMID 27017469.
- ↑ 23.0 23.1 23.2 D'Antonio LL, Scherer NJ, Miller LL, Kalbfleisch JH, Bartley JA (2001). "Analysis of speech characteristics in children with velocardiofacial syndrome (VCFS) and children with phenotypic overlap without VCFS". Cleft Palate Craniofac. J. 38 (5): 455–67. doi:10.1597/1545-1569(2001)038<0455:AOSCIC>2.0.CO;2. ISSN 1545-1569. PMID 11522167.
- ↑ 24.0 24.1 24.2 Scherer NJ, D'Antonio LL, Kalbfleisch JH (1999). "Early speech and language development in children with velocardiofacial syndrome". Am. J. Med. Genet. 88 (6): 714–23. doi:10.1002/(SICI)1096-8628(19991215)88:6<714::AID-AJMG24>3.0.CO;2-B. PMID 10581495.
- ↑ Scherer NJ, D'Antonio LL, Rodgers JR (2001). "Profiles of communication disorder in children with velocardiofacial syndrome: comparison to children with Down syndrome". Genet. Med. 3 (1): 72–8. doi:10.1097/00125817-200101000-00016. PMID 11339384.
- ↑ Eliez S, Palacio-Espasa F, Spira A (2000). "Young children with Velo-Cardio-Facial syndrome (CATCH-22). Psychological and language phenotypes". Eur Child Adolesc Psychiatry. 9 (2): 109–14. doi:10.1007/s007870050005. PMID 10926060.
- ↑ 27.0 27.1 27.2 Robin NH, Shprintzen RJ (2005). "Defining the clinical spectrum of deletion 22q11.2". J. Pediatr. 147 (1): 90–6. doi:10.1016/j.jpeds.2005.03.007. PMID 16027702.
- ↑ 28.0 28.1 Solot CB, Knightly C, Handler SD (2000). "Communication disorders in the 22Q11.2 microdeletion syndrome". J Commun Disord. 33 (3): 187–203, quiz 203–4. doi:10.1016/S0021-9924(00)00018-6. PMID 10907715.
- ↑ Persson C, Niklasson L, Oskarsdóttir S, Johansson S, Jönsson R, Söderpalm E (2006). "Language skills in 5-8-year-old children with 22q11 deletion syndrome". Int J Lang Commun Disord. 41 (3): 313–33. doi:10.1080/13682820500361497. PMID 16702096.
- ↑ 30.0 30.1 30.2 Online Mendelian Inheritance in Man (OMIM): #188400
- ↑ 31.0 31.1 Packham EA, Brook JD (April 2003). "T-box genes in human disorders". Hum. Mol. Genet. 12 Spec No 1 (90001): R37–44. doi:10.1093/hmg/ddg077. PMID 12668595.
- ↑ Tang KL, Antshel KM, Fremont WP, Kates WR (October 2015). "Behavioral and Psychiatric Phenotypes in 22q11.2 Deletion Syndrome". J Dev Behav Pediatr. 36 (8): 639–50. doi:10.1097/DBP.0000000000000210. PMC 4586411. PMID 26372046.
- ↑ Maynard TM, Meechan DW, Dudevoir ML, Gopalakrishna D, Peters AZ, Heindel CC, Sugimoto TJ, Wu Y, Lieberman JA, Lamantia AS (November 2008). "Mitochondrial localization and function of a subset of 22q11 deletion syndrome candidate genes". Mol. Cell. Neurosci. 39 (3): 439–51. doi:10.1016/j.mcn.2008.07.027. PMC 2729512. PMID 18775783.
- ↑ 34.0 34.1 Bartsch O, Nemecková M, Kocárek E, Wagner A, Puchmajerová A, Poppe M, Ounap K, Goetz P (February 2003). "DiGeorge/velocardiofacial syndrome: FISH studies of chromosomes 22q11 and 10p14, and clinical reports on the proximal 22q11 deletion". Am. J. Med. Genet. A. 117A (1): 1–5. doi:10.1002/ajmg.a.10914. PMID 12548732.
- ↑ Poirsier C, Besseau-Ayasse J, Schluth-Bolard C, Toutain J, Missirian C, Le Caignec C, et al. (June 2016). "A French multicenter study of over 700 patients with 22q11 deletions diagnosed using FISH or aCGH". Eur. J. Hum. Genet. 24 (6): 844–51. doi:10.1038/ejhg.2015.219. PMC 4867458. PMID 26508576.
- ↑ Edelmann L, Pandita RK, Spiteri E, Funke B, Goldberg R, Palanisamy N, Chaganti RS, Magenis E, Shprintzen RJ, Morrow BE (1999). "A common molecular basis for rearrangement disorders on chromosome 22q11". Hum Mol Genet. 8 (7): 1157–67. doi:10.1093/hmg/8.7.1157. PMID 10369860. Archived from the original on 2021-04-22. Retrieved 2015-11-11.
- ↑ Jerome LA, Papaioannou VE (March 2001). "DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1". Nat. Genet. 27 (3): 286–91. doi:10.1038/85845. PMID 11242110.
- ↑ Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, Bradley A, Baldini A (March 2001). "Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice". Nature. 410 (6824): 97–101. doi:10.1038/35065105. PMID 11242049.
- ↑ Chun S, Du F, Westmoreland JJ, Han SB, Wang YD, Eddins D, et al. (January 2017). "Thalamic miR-338-3p mediates auditory thalamocortical disruption and its late onset in models of 22q11.2 microdeletion". Nat. Med. 23 (1): 39–48. doi:10.1038/nm.4240. PMC 5218899. PMID 27892953.
- ↑ "Gene (NCBI)". Archived from the original on 2021-04-16. Retrieved 2020-07-11.
- ↑ 41.0 41.1 Kremer LS, Distelmaier F, Alhaddad B, Hempel M, Iuso A, Küpper C, et al. (2016). "Bi-allelic Truncating Mutations in TANGO2 Cause Infancy-Onset Recurrent Metabolic Crises with Encephalocardiomyopathy". American Journal of Human Genetics. 98 (2): 358–62. doi:10.1016/j.ajhg.2015.12.009. PMC 4746337. PMID 26805782.
- ↑ 42.0 42.1 Lalani SR, Liu P, Rosenfeld JA, Watkin LB, Chiang T, Leduc MS, et al. (2016). "Recurrent Muscle Weakness with Rhabdomyolysis, Metabolic Crises, and Cardiac Arrhythmia Due to Bi-allelic TANGO2 Mutations". American Journal of Human Genetics. 98 (2): 347–57. doi:10.1016/j.ajhg.2015.12.008. PMC 4746334. PMID 26805781.
- ↑ Miller, Kimberley A. (2008). "FISH Diagnosis of 22q11.2 Deletion Syndrome". Newborn and Infant Nursing Reviews. 8 (1): e11–e19. doi:10.1053/j.nainr.2007.12.006.
- ↑ Jalali GR, Vorstman JA, Errami A, Vijzelaar R, Biegel J, Shaikh T, Emanuel BS (March 2008). "Detailed analysis of 22q11.2 with a high density MLPA probe set". Hum. Mutat. 29 (3): 433–40. doi:10.1002/humu.20640. PMC 2664158. PMID 18033723.
- ↑ García-Herrero S, Campos-Galindo I, Martínez-Conejero JA, Serra V, Olmo I, Lara C, Simón C, Rubio C (2014). "BACs-on-Beads technology: a reliable test for rapid detection of aneuploidies and microdeletions in prenatal diagnosis". Biomed Res Int. 2014: 590298. doi:10.1155/2014/590298. PMC 3985206. PMID 24795887.
- ↑ Choy KW, Kwok YK, Cheng YK, Wong KM, Wong HK, Leung KO, Suen KW, Adler K, Wang CC, Lau TK, Schermer MJ, Lao TT, Leung TY (September 2014). "Diagnostic accuracy of the BACs-on-Beads™ assay versus karyotyping for prenatal detection of chromosomal abnormalities: a retrospective consecutive case series". BJOG. 121 (10): 1245–52. doi:10.1111/1471-0528.12873. PMID 24893808.
- ↑ Park SJ, Jung EH, Ryu RS, Kang HW, Ko JM, Kim HJ, Cheon CK, Hwang SH, Kang HY (May 2011). "Clinical implementation of whole-genome array CGH as a first-tier test in 5080 pre and postnatal cases". Mol Cytogenet. 4: 12. doi:10.1186/1755-8166-4-12. PMC 3114015. PMID 21549014.
- ↑ Mupanemunda, Richard H.; Watkinson, Michael (2004). Key Topics in Neonatology. CRC Press. p. 82. ISBN 9781859962343.
- ↑ Tonelli AR, Kosuri K, Wei S, Chick D (2007). "Seizures as the first manifestation of chromosome 22q11.2 deletion syndrome in a 40-year old man: a case report". J Med Case Rep. 1: 167. doi:10.1186/1752-1947-1-167. PMC 2222674. PMID 18053182.
- ↑ "DiGeorge (22q11.2 deletion) syndrome: Management and prognosis". www.uptodate.com. Archived from the original on 2018-10-31. Retrieved 2018-10-30.
- ↑ Markert ML, Devlin BH, Alexieff MJ, Li J, McCarthy EA, Gupton SE, et al. (May 2007). "Review of 54 patients with complete DiGeorge anomaly enrolled in protocols for thymus transplantation: outcome of 44 consecutive transplants". Blood. 109 (10): 4539–47. doi:10.1182/blood-2006-10-048652. PMC 1885498. PMID 17284531.
- ↑ "Clinical and Metabolic Genetics- The 22q Deletion Clinic". The Hospital for Sick Children. Archived from the original on 2016-04-07.
- ↑ Fung WL, Butcher NJ, Costain G, Andrade DM, Boot E, Chow EW, et al. (August 2015). "Practical guidelines for managing adults with 22q11.2 deletion syndrome". Genet. Med. 17 (8): 599–609. doi:10.1038/gim.2014.175. PMC 4526275. PMID 25569435.
- ↑ Oskarsdóttir S, Vujic M, Fasth A (2004). "Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in Western Sweden". Arch. Dis. Child. 89 (2): 148–51. doi:10.1136/adc.2003.026880. PMC 1719787. PMID 14736631.
- ↑ Daily DK, Ardinger HH, Holmes GE (February 2000). "Identification and evaluation of mental retardation". Am Fam Physician. 61 (4): 1059–67, 1070. PMID 10706158.
- ↑ "The Genetics of 22q11.2 DS: Demographics". Information for Medical Professionals. The Dalglish Family Hearts and Minds Clinic for Adults with 22q11.2 Deletion Syndrome. Archived from the original on 9 March 2016. Retrieved 26 August 2015.
- ↑ "Di George's syndrome". 2015 ICD-10-CM Diagnosis Code D82.1. Archived from the original on 24 September 2015. Retrieved 26 August 2015.
- ↑ 58.0 58.1 "Velo-cardio-facial syndrome". 2015 ICD-10-CM Diagnosis Code Q93.81. Archived from the original on 24 September 2015. Retrieved 26 August 2015.
- ↑ Bassett AS, McDonald-McGinn DM, Devriendt K, Digilio MC, Goldenberg P, Habel A, Marino B, Oskarsdottir S, Philip N, Sullivan K, Swillen A, Vorstman J (August 2011). "Practical guidelines for managing patients with 22q11.2 deletion syndrome". J. Pediatr. 159 (2): 332–9.e1. doi:10.1016/j.jpeds.2011.02.039. PMC 3197829. PMID 21570089.
- ↑ "Same Name Campaign - 22q.org". 22q.org. Archived from the original on 2017-06-10. Retrieved 2017-06-18.
This article incorporates public domain text from The U.S. National Library of Medicine Archived 2019-02-04 at the Wayback Machine
External links
| Classification | |
|---|---|
| External resources |
- DiGeorge syndrome at Curlie
- McDonald-McGinn DM, Emanuel BS, Zackai EH (December 16, 2005). "22q11.2 Deletion Syndrome". In Pagon RA, Bird TD, Dolan CR, Stephens K (eds.). GeneReviews. PMID 20301696. NBK1523.
{{cite book}}:|access-date=requires|url=(help);|archive-url=requires|url=(help); Unknown parameter|chapterurl=ignored (help) - Firth HV (February 17, 2009). "22q11.2 Duplication". In Pagon RA, Bird TD, Dolan CR, Stephens K (eds.). GeneReviews. PMID 20301749. NBK3823.
{{cite book}}:|access-date=requires|url=(help);|archive-url=requires|url=(help); Unknown parameter|chapterurl=ignored (help)
| Main topics | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Approaches | |||||||||||
| Rights, law, support |
| ||||||||||
| Structural and assistive | |||||||||||
| Social issues | |||||||||||
| Disabilities | |||||||||||
| Arts, media, culture, sport | |||||||||||
- Pages with script errors
- All pages needing factual verification
- Wikipedia articles needing factual verification from April 2015
- Articles with invalid date parameter in template
- CS1: long volume value
- Articles contradicting other articles
- All articles with unsourced statements
- Articles with unsourced statements from June 2017
- Webarchive template wayback links
- Articles with Curlie links
- CS1 errors: unsupported parameter
- CS1 errors: access-date without URL
- CS1 errors: archive-url
- Autosomal dominant disorders
- IUIS-PID table 3 immunodeficiencies
- Noninfectious immunodeficiency-related cutaneous conditions
- Syndromes affecting the heart
- Autosomal monosomies and deletions
- Chromosomal abnormalities
- RTT
- Syndromes with craniofacial abnormalities
- Medical mnemonics