Tuberous sclerosis
| Tuberous sclerosis | |
|---|---|
| Other names: Tuberous sclerosis complex (TSC); Bourneville Pringle syndrome[1] | |
 | |
| A case of tuberous sclerosis showing facial angiofibromas in the characteristic butterfly pattern | |
| Pronunciation |
|
| Specialty | Neurology, medical genetics |
| Symptoms | Seizures, intellectual disability, developmental delay, behavioral problems, autism[3] |
| Complications | Kidney and lung problems[3] |
| Duration | Life-long[3] |
| Causes | Genetic disorder (autosomal dominant)[3] |
| Diagnostic method | Based on symptoms, supported by medical imaging[3] |
| Differential diagnosis | Focal cortical dysplasia[4] |
| Treatment | Physical therapy, occupational therapy, speech therapy, behavioral therapy, antiepileptic medication[3] |
| Prognosis | Variable[3] |
| Frequency | 1 in 6,000[3] |
Tuberous sclerosis (TS) is a genetic disorder that results in the growth of non-cancerous tumours in the brain and other organs such as the heart, eyes, lungs, and skin.[3] Common symptoms include seizures, intellectual disability, developmental delay, behavioral problems, and autism.[3] Onset of problems is often in the first year of life; though may occur later.[3] Complications may include kidney and lung problems.[3]
It is caused by a mutation of either the TSC1 or TSC2 gene, which code for the protein hamartin and tuberin, respectively.[3] While it may be inherited from a person's parents in an autosomal dominant manner, most cases occur as new mutations during early development.[3] These proteins are believed to be tumor growth suppressors.[3] Diagnosis is generally based on symptoms and supported by medical imaging.[3] It may also be confirmed by genetic testing.[5]
There is no cure, with treatment being supportive in nature.[3] Efforts may include physical therapy, occupational therapy, speech therapy, and behavioral therapy.[3] Antiepileptic medication, such as vigabatrin, may be used for seizures.[3] Long-term outcomes are variable and depends on the severity of the symptoms.[3]
Tuberous sclerosis may affect up to 2 million people worldwide.[1] In the United States it affects about 1 in 6,000 children; while it is a somewhat less common in Europe.[3][1] Males and females are affected with similar frequency.[1] It was first described by French neurologist Désiré-Magloire Bourneville in 1880.[6] The name refers to the irregular swellings in the brains, that maybe shaped like potatoes.[3]
Signs and symptoms

The manifestations are due to the formation of hamartia (malformed tissue such as the cortical tubers), hamartomas (benign growths such as facial angiofibroma and subependymal nodules), and very rarely, cancerous hamartoblastomas. The effect of these on the brain leads to neurological symptoms such as seizures, intellectual disability, developmental delay, and behavioral problems.
Individuals may experience variable numbers of symptoms.
Skin
Some form of skin problem is present in 96% of individuals. Most cause no problems, but are helpful in diagnosis. Some cases may cause disfigurement, necessitating treatment. The most common skin abnormalities include:
- Hypomelanic macules ("ash leaf spots") are present in about 90% of people with TSC.[8] These small white or lighter patches of skin may appear anywhere on the body, and are caused by a lack of melanin. They are usually the only visible sign of TSC at birth. In fair-skinned individuals, a Wood's lamp (ultraviolet light) may be required to see them. On the scalp, the effect may be a white patch of hair (poliosis). Patches smaller than 3mm are known as "confetti" skin lesions.[8]
- Facial angiofibromas are present in about 75% of people with TSC.[8] These are a rash of reddish spots or bumps on the nose and cheeks in a butterfly distribution, which consist of blood vessels and fibrous tissue. This potentially socially embarrassing rash starts to appear during childhood.
- Ungual fibromas: Also known as Koenen's tumors, these are small fleshy tumors that grow around and under the toenails or fingernails. These are rare in childhood, but common by middle age.[9] They are generally more common on toes than on fingers, develop at 15–29 years, and are more common in women than in men.
- Fibrous cephalic plaques are present in about 25% of people with TSC.[8] These are raised, discoloured areas usually found on the forehead, but sometimes on the face or elsewhere on the scalp.
- Shagreen patches are present in about half of people with TSC, appearing in childhood.[8] They are areas of thick leathery skin that are dimpled like an orange peel, and pigmented, they are usually found on the lower back or nape of the neck, or scattered across the trunk or thighs. The frequency of these lesions rises with age.
- Dental enamel pits are found in almost all adults with TSC.[8]
- Intraoral fibromas are small surface-tumours found in the gums, inside the cheeks or tongue. Gum (gingival) fibromas are found in about 20-50% of people with TSC, more commonly in adults.[8]
-
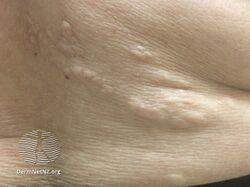
Tuberous sclerosis shagreen patch
-

Tuberous sclerosis digital fibromas
-
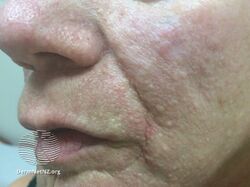
Tuberous sclerosis angiofibromas
.jpg)
.jpg)
.jpg)
Neurological

Three types of brain tumours are associated with TSC:
- Giant cell astrocytoma: (grows and blocks the cerebrospinal fluid flow, leading to dilatation of ventricles causing headache and vomiting)
- Cortical tubers: after which the disease is named
- Subependymal nodules: form in the walls of ventricles
Classic intracranial manifestations of TSC include subependymal nodules and cortical/subcortical tubers.[10]
The tubers are typically triangular in configuration, with the apex pointed towards the ventricles, and are thought to represent foci of abnormal neuronal migration. The T2 signal abnormalities may subside in adulthood, but will still be visible on histopathological analysis. On magnetic resonance imaging (MRI), TSC patients can exhibit other signs consistent with abnormal neuron migration such as radial white matter tracts hyperintense on T2WI and heterotopic gray matter.[citation needed]
Subependymal nodules are composed of abnormal, swollen glial cells and bizarre multinucleated cells which are indeterminate for glial or neuronal origin. Interposed neural tissue is not present. These nodules have a tendency to calcify as the patient ages. A nodule that markedly enhances and enlarges over time should be considered suspicious for transformation into a subependymal giant cell astrocytoma, which typically develops in the region of the foramen of Monro, in which case it is at risk of developing an obstructive hydrocephalus.[citation needed]
A variable degree of ventricular enlargement is seen, either obstructive (e.g. by a subependymal nodule in the region of the foramen of Monro) or idiopathic in nature.[citation needed]
Neuropsychiatric
About 90% of people with TSC develop a range of neurodevelopmental, behavioural, psychiatric, and psychosocial difficulties. The "TSC‐associated neuropsychiatric disorders" are abbreviated TAND. These difficulties are less frequently identified and thus undertreated when compared with the neurological symptoms.[11] Most problems are associated with more severe intellectual delay or associated with childhood and adolescence, and some (for example depressed mood) may be unreported if the person is unable to communicate. TAND can be investigated and considered at six levels: behavioural, psychiatric, intellectual, academic, neuropsychological, and psychosocial.[11]
Behavioural problems most commonly seen include overactivity, impulsivity and sleeping difficulties. Also common are anxiety, mood swings, and severe aggression. Less common are depressed mood, self-injury, and obsessional behaviours.[11]
People with TSC are frequently also diagnosed with psychiatric disorders: autism spectrum disorder (ASD), attention deficit hyperactivity disorder (ADHD), anxiety disorder and depressive disorder. TSC is one of the most common genetic causes of autism spectrum disorder, which affects nearly half of people with TSC. ASD is more common in TSC2 than TSC1 and more common with earlier and more severe epilepsy, and with lower intellectual ability. ADHD is nearly as frequently seen in TSC as ASD (up to half of all people with TSC). Anxiety and depressive disorders, when they occur, are typically diagnosed in early adulthood and among those intellectually able to express their moods.[11] Schizophrenia (and symptoms like hallucinations or psychosis) is no more common in TSC than the general population.[citation needed]
The intellectual ability of people with TSC varies enormously. About 40–50% have a normal IQ. A normal IQ is much more commonly seen in TSC1 than TSC2, and profound intellectual disability seen in 34% of TSC2 compared with 10% of TSC1 in one study. Many studies have examined whether early onset, type and severity of epilepsy associates with intellectual ability. Academic issues occur even in people with TSC who have normal intellectual ability. These are often specific learning disorders such as dyscalculia (understanding mathematics), but also include other aspects affecting school life such as anxiety, lack of social skills or low self-esteem.[11]
About half of people with TSC, when assessed for neuropsychological skills, are in the bottom 5th percentile in some areas, which indicates a severe impairment. These include problems with attention (for example, being able to concentrate on two separate things like looking and listening), memory (particularly recall, verbal and spatial working memory) and executive function (for example, planning, self-monitoring, cognitive flexibility).[11]
The psychosocial impacts of TSC include low self-esteem and self-efficacy in the individual, and a burden on the family coping with a complex and unpredictable disorder.[11]
Kidneys

Between 60 and 80% of people with tuberous sclerosis have benign tumors (once thought hamartomatous, but now considered true neoplasms) of the kidneys called angiomyolipomas frequently causing hematuria. These tumors are composed of vascular (angio–), smooth muscle (–myo–), and fat (–lip-) tissue. Although benign, an angiomyolipoma larger than 4 cm is at risk for a potentially catastrophic hemorrhage either spontaneously or with minimal trauma. Angiomyolipomas are found in about one in 300 people without TSC. However, those are usually solitary, whereas in TSC they are commonly multiple and bilateral.
About 20-30% of people with TSC have renal cysts, causing few problems. However, 2% may also have autosomal dominant polycystic kidney disease.
Very rare (< 1%) problems include renal cell carcinoma and oncocytomas (benign adenomatous hamartoma).
Lungs
Patients with TSC can develop progressive replacement of the lung parenchyma with multiple cysts, known as lymphangioleiomyomatosis (LAM). Recent genetic analysis has shown that the proliferative bronchiolar smooth muscle in TSC-related lymphangioleiomyomatosis is monoclonal metastasis from a coexisting renal angiomyolipoma. Cases of TSC-related lymphangioleiomyomatosis recurring following lung transplant have been reported.[12]
Heart
Small tumours of the heart muscle, called cardiac rhabdomyomas, are rare in the general population (perhaps 0.2% of children) but very common in people with TSC. Around 80% of children under two-years-old with TSC have at least one rhabdomyoma, and about 90% of those will have several. The vast majority of children with at least one rhabdomyoma, and nearly all children with multiple rhabdomyomas will be found to have TSC. Prenatal ultrasound, performed by an obstetric sonographer specializing in cardiology, can detect a rhabdomyoma after 20 weeks. Rhabdomyoma vary in size from a few millimetres to several centimetres, and are usually found in the lower chambers (ventricles) and less often in the upper chambers (atria). They grow in size during the second half of pregnancy, but regress after birth, and are seen in only around 20% of children over two years old.[13]
Most rhabdomyomas cause no problems but some may cause heart failure in the foetus or first year of life. Rhabdomyomas are believed to be responsible for the development of heart arrhythmia later in life, which is relatively common in TSC. Arrhythmia can be hard to spot in people with TSC, other than by performing routine ECG. For example, arrhythmia may cause fainting that is confused with drop seizures, and symptoms of arrhythmia such as palpitations may not be reported in an individual with developmental delay.[13]
Eyes
Retinal lesions, called astrocytic hamartomas (or "phakomas"), which appear as a greyish or yellowish-white lesion in the back of the globe on the ophthalmic examination. Astrocytic hamartomas can calcify, and they are in the differential diagnosis of a calcified globe mass on a CT scan.[14]
Nonretinal lesions associated with TSC include:
- Coloboma[14]
- Angiofibromas of the eyelids[14]
- Papilledema (related to hydrocephalus)
Pancreas
Pancreatic neuroendocrine tumours have been described in rare cases of TSC.[15]
Genetics

TSC is a genetic disorder with an autosomal dominant pattern of inheritance, variable expressivity, and incomplete penetrance.[9][16] Two-thirds of TSC cases result from sporadic genetic mutations, not inheritance, but their offspring may inherit it from them. Current genetic tests have difficulty locating the mutation in roughly 20% of individuals diagnosed with the disease. So far, it has been mapped to two genetic loci, TSC1 and TSC2.[citation needed]
TSC1 encodes for the protein hamartin, is located on chromosome 9 q34, and was discovered in 1997.[17] TSC2 encodes for the protein tuberin, is located on chromosome 16 p13.3, and was discovered in 1993.[18] TSC2 is contiguous with PKD1, the gene involved in one form of polycystic kidney disease (PKD). Gross deletions affecting both genes may account for the 2% of individuals with TSC who also develop polycystic kidney disease in childhood.[19] TSC2 has been associated with a more severe form of TSC.[20] However, the difference is subtle and cannot be used to identify the mutation clinically. Estimates of the proportion of TSC caused by TSC2 range from 55% to 90%.[21]
TSC1 and TSC2 are both tumor suppressor genes that function according to Knudson's "two hit" hypothesis. That is, a second random mutation must occur before a tumor can develop. This explains why, despite its high penetrance, TSC has wide expressivity.[citation needed]
Column-generating template families
The templates listed here are not interchangeable. For example, using {{col-float}} with {{col-end}} instead of {{col-float-end}} would leave a HTML "div" (division) open, potentially harming any subsequent formatting. <section begin="table" />
| Type | Family | Handles wiki
table code?† |
Responsive/ Mobile suited |
Start template | Column divider | End template |
|---|---|---|---|---|---|---|
| Float | "col-float" | Yes | Yes | {{col-float}} | {{col-float-break}} | {{col-float-end}} |
| "columns-start" | Yes | Yes | {{columns-start}} | {{column}} | {{columns-end}} | |
| Columns | "div col" | Yes | Yes | {{div col}} | – | {{div col end}} |
| "columns-list" | No | Yes | {{columns-list}} (wraps div col) | – | – | |
| Flexbox | "flex columns" | No | Yes | {{flex columns}} | – | – |
| Table | "col" | Yes | No | {{col-begin}}, {{col-begin-fixed}} or {{col-begin-small}} |
{{col-break}} or {{col-2}} .. {{col-5}} |
{{col-end}} |
† Can template handle the basic wiki markup {| | || |- |} used to create tables? If not, special templates that produce these elements (such as {{(!}}, {{!}}, {{!!}}, {{!-}}, {{!)}})—or HTML tags (<table>...</table>, <tr>...</tr>, etc.)—need to be used instead.<section end="table" />
Pathophysiology
Hamartin and tuberin function as a complex which is involved in the control of cell growth and cell division. The complex appears to interact with RHEB GTPase, thus sequestering it from activating mTOR signalling, part of the growth factor (insulin) signalling pathway. Thus, mutations at the TSC1 and TSC2 loci result in a loss of control of cell growth and cell division, and therefore a predisposition to forming tumors. TSC affects tissues from different germ layers. Cutaneous and visceral lesions may occur, including angiofibroma, cardiac rhabdomyomas, and renal angiomyolipomas. The central nervous system lesions seen in this disorder include hamartomas of the cortex, hamartomas of the ventricular walls, and subependymal giant cell tumors, which typically develop in the vicinity of the foramina of Monro.[citation needed]
Molecular genetic studies have defined at least two loci for TSC. In TSC1, the abnormality is localized on chromosome 9q34, but the nature of the gene protein, called hamartin, remains unclear. No missense mutations occur in TSC1. In TSC2, the gene abnormalities are on chromosome 16p13. This gene encodes tuberin, a guanosine triphosphatase–activating protein. The specific function of this protein is unknown. In TSC2, all types of mutations have been reported; new mutations occur frequently. Few differences have yet been observed in the clinical phenotypes of patients with mutation of one gene or the other.[citation needed]
Cells from individuals with pathogenic mutations in the TSC2 gene display abnormal accumulation of glycogen that is associated with depletion of lysosomes and autophagic impairment. The defective degradation of glycogen by the autophagy-lysosome pathway is, at least in part, independent of impaired regulation of mTORC1 and is restored, in cultured cells, by the combined use of PKB/Akt and mTORC1 pharmacological inhibitors.[22]
Diagnosis
Tuberous sclerosis complex is diagnosed with clinical and genetic tests. There are many different mutations in the TSC1 and TSC2 genes that have been identified in individuals with TSC. A pathogenic mutation in the gene prevents the proteins from being made or inactivates the proteins. If such a pathogenic mutation is found then this alone is sufficient to diagnose TSC. However, some mutations are less clear in their effect, and so not sufficient alone for diagnosis. Between 1 in 10 and 1 in 4 of individuals with TSC have no mutation that can be identified. Once a particular mutation is identified in someone with TSC, this mutation can be used to make confident diagnoses in other family members.[8]
For clinical diagnosis, there isn't one sign that is unique (pathognomonic) to TSC, nor are all signs seen in all individuals. Therefore, several signs are considered together, classed as either major or minor features. An individual with two major features, or one major feature and at least two minor features can be given a definite diagnosis of TSC. If only one major feature or at least two minor features are present, the diagnosis is only regarded as possibly TSC.[8]
| Major Features | ||||
|---|---|---|---|---|
| Location | Sign | Onset[23] | Note | |
| 1 | Skin | Hypomelanotic macules | Infant – child | At least three, at least 5 mm in diameter. |
| 2 | Head | Facial angiofibromas or fibrous cephalic plaque | Infant – adult | At least three angiofibromas |
| 3 | Fingers and toes | Ungual fibroma | Adolescent – adult | At least two |
| 4 | Skin | Shagreen patch (connective tissue nevus) | Child | |
| 5 | Eyes | Multiple retinal nodular hamartomas | Infant | |
| 6 | Brain | Cortical dysplasias (includes tubers and cerebral white matter radial migration lines) | Fetus | |
| 7 | Brain | Subependymal nodule | Child – adolescent | |
| 8 | Brain | Subependymal giant cell astrocytoma | Child – adolescent | |
| 9 | Heart | Cardiac rhabdomyoma | Fetus | |
| 10 | Lungs | Lymphangioleiomyomatosis | Adolescent – adult | |
| 11 | Kidneys | Renal angiomyolipoma | Child – adult | At least two. Together, 10 and 11 count as one major feature. |
| Minor Features | ||||
| Location | Sign | Note | ||
| 1 | Skin | "Confetti" skin lesions | ||
| 2 | Teeth | Dental enamel pits | At least three | |
| 3 | Gums | Intraoral fibromas | At least two | |
| 4 | Eyes | Retinal achromic patch | ||
| 5 | Kidneys | Multiple renal cysts | ||
| 6 | Liver, spleen and other organs | Nonrenal hamartoma | ||
TSC can be first diagnosed at any stage of life. Prenatal diagnosis is possible by chance if heart tumours are discovered during routine ultrasound. In infancy, epilepsy, particularly infantile spasms, or developmental delay may lead to neurological tests. The white patches on the skin may also first become noticed. In childhood, behavioural problems and autism spectrum disorder may provoke a diagnosis. During adolescence, the skin problems appear. In adulthood, kidney and lung problems may develop. An individual may also be diagnosed at any time as a result of genetic testing of family members of another affected person.[24]
Management
Tuberous sclerosis complex affects multiple organ systems so a multidisciplinary team of medical professionals is required.[citation needed]
In suspected or newly diagnosed TSC, the following tests and procedures are recommended by 2012 International Tuberous Sclerosis Complex Consensus Conference.[25]
- Take a personal and family history covering three generations. Genetic counselling and tests determine if other individuals are at risk.
- A magnetic resonance imaging (MRI) of the brain to identify tubers, subependymal nodules (SEN) and sub-ependymal giant cell astrocytomas (SEGA).
- Children undergo a baseline electroencephalograph (EEG) and family educated to identify seizures if/when they occur.
- Assess children for behavioural issues, autism spectrum disorder, psychiatric disorders, developmental delay, and neuropsychological problems.
- Scan the abdomen for tumours in various organs, but most importantly angiomyolipomata in the kidneys. MRI is superior to CT or ultrasound. Take blood pressure and test renal function.
- In adult women, test pulmonary function and perform a high-resolution computed tomography (HRCT) of the chest.
- Examine the skin under a Wood's lamp (hypomelanotic macules), the fingers and toes (ungual fibroma), the face (angiofibromas), and the mouth (dental pits and gingival fibromas).
- In infants under three, perform an echocardiogram to spot rhabdomyomas, and electrocardiogram (ECG) for any arrhythmia.
- Use a fundoscope to spot retinal hamartomas or achromic patches.
The various symptoms and complications from TSC may appear throughout life, requiring continued surveillance and adjustment to treatments. The following ongoing tests and procedures are recommended by 2012 International Tuberous Sclerosis Complex Consensus Conference.[25]
- In children and adults younger than 25 years, a magnetic resonance imaging (MRI) of the brain is performed every one to three years to monitor for subependymal giant cell astrocytoma (SEGA). If a SEGA is large, growing or interfering with ventricles, the MRI is performed more frequently. After 25 years, if there are no SEGAs then periodic scans may no longer be required. A SEGA causing acute symptoms are removed with surgery, otherwise either surgery or drug treatment with an mTOR inhibitor may be indicated.
- Repeat screening for TSC-associated neuropsychiatric disorders (TAND) at least annually. Sudden behavioural changes may indicate a new physical problem (for example with the kidneys, epilepsy or a SEGA).
- Routine EEG determined by clinical need.
- Infantile spasms are best treated with vigabatrin and adrenocorticotropic hormone used as a second-line therapy. Other seizure types have no TSC-specific recommendation, though epilepsy in TSC is typically difficult to treat (medically refractory).
- Repeat MRI of abdomen every one to three years throughout life. Check renal (kidney) function annually. Should angiomyolipoma bleed, this is best treated with embolisation and then corticosteroids. Removal of the kidney (nephrectomy) is strongly to be avoided. An asymptomatic angiomyolipoma that is growing larger than 3 cm is best treated with an mTOR inhibitor drug. Other renal complications spotted by imaging include polycystic kidney disease and renal cell carcinoma.
- Repeat chest HRCT in adult women every five to 10 years. Evidence of lymphangioleiomyomatosis (LAM) indicates more frequent testing. An mTOR inhibitor drug can help, though a lung transplant may be required.
- A 12-lead ECG should be performed every three to five years.
The mTOR inhibitor everolimus was approved in the US for treatment of TSC-related tumors in the brain (subependymal giant cell astrocytoma) in 2010 and in the kidneys (renal angiomyolipoma) in 2012.[26][27] Oral everolimus (rapalog) reduces tumour size, is effective in terms of response to skin lesions and does not increase the risk of adverse events.[28] Everolimus also showed evidence of effectiveness at treating epilepsy in some people with TSC.[29][30] In 2017, the European Commission approved everolimus for treatment of refractory partial-onset seizures associated with TSC.[31]
Neurosurgical intervention may reduce the severity and frequency of seizures in TSC patients.[32][33] Embolization and other surgical interventions can be used to treat renal angiomyolipoma with acute hemorrhage. Surgical treatments for symptoms of lymphangioleiomyomatosis (LAM) in adult TSC patients include pleurodesis to prevent pneumothorax and lung transplantation in the case of irreversible lung failure.[25]
Other treatments that have been used to treat TSC manifestations and symptoms include a ketogenic diet for intractable epilepsy and pulmonary rehabilitation for LAM.[34] Facial angiofibromas can be reduced with laser treatment and the effectiveness of mTOR inhibitor topical treatment is being investigated. Laser therapy is painful, requires anaesthesia, and has risks of scarring and dyspigmentation.[35]
Prognosis
The prognosis for individuals with TSC depends on the severity of symptoms, which range from mild skin abnormalities to varying degrees of learning disabilities and epilepsy to severe intellectual disability, uncontrollable seizures, and kidney failure. Those individuals with mild symptoms generally do well and live long, productive lives, while individuals with the more severe form may have serious disabilities. However, with appropriate medical care, most individuals with the disorder can look forward to normal life expectancy.[36]
A study of 30 TSC patients in Egypt found, "...earlier age of seizures commencement (<6 months) is associated with poor seizure outcome and poor intellectual capabilities. Infantile spasms and severely epileptogenic EEG patterns are related to the poor seizure outcome, poor intellectual capabilities and autistic behavior. Higher tubers numbers is associated with poor seizure outcome and autistic behavior. Left-sided tuber burden is associated with poor intellect, while frontal location is more encountered in ASD [autism spectrum disorders]. So, close follow up for the mental development and early control of seizures are recommended in a trial to reduce the risk factors of poor outcome. Also early diagnosis of autism will allow for earlier treatment and the potential for better outcome for children with TSC."[37]
Leading causes of death include renal disease, brain tumour, lymphangioleiomyomatosis of the lung, and status epilepticus or bronchopneumonia in those with severe mental handicap.[38] Cardiac failure due to rhabdomyomas is a risk in the fetus or neonate but is rarely a problem subsequently. Kidney complications such as angiomyolipoma and cysts are common and more frequent in females than males and in TSC2 than TSC1. Renal cell carcinoma is uncommon. Lymphangioleiomyomatosis is only a risk for females with angiomyolipomas.[39] In the brain, the subependymal nodules occasionally degenerate to subependymal giant cell astrocytomas. These may block the circulation of cerebrospinal fluid around the brain, leading to hydrocephalus.[citation needed]
Detection of the disease should be followed by genetic counselling. It is also important to realise that though the disease does not have a cure, symptoms can be treated symptomatically. Hence, awareness regarding different organ manifestations of TSC is important.[citation needed]
Epidemiology
TSC occurs in all races and ethnic groups, and in both genders. The live-birth prevalence is estimated to be between 10 and 16 cases per 100,000. A 1998 study[40] estimated total population prevalence between about 7 and 12 cases per 100,000, with more than half of these cases undetected. Prior to the invention of CT scanning to identify the nodules and tubers in the brain, the prevalence was thought to be much lower, and the disease associated with those people diagnosed clinically with learning disability, seizures and facial angiofibroma. Whilst still regarded as a rare disease, TSC is common when compared to many other genetic diseases, with at least 1 million individuals affected worldwide.[7]
History

TSC first came to medical attention when dermatologists described the distinctive facial rash (1835 and 1850). A more complete case was presented by von Recklinghausen (1862), who identified heart and brain tumours in a newborn who had only briefly lived. However, Bourneville (1880) is credited with having first characterized the disease, coining the name "tuberous sclerosis", thus earning the eponym Bourneville's disease. The neurologist Vogt (1908) established a diagnostic triad of epilepsy, idiocy, and adenoma sebaceum (an obsolete term for facial angiofibroma).[41]
Symptoms were periodically added to the clinical picture. The disease as presently understood was first fully described by Gomez (1979). The invention of medical ultrasound, CT and MRI has allowed physicians to examine the internal organs of live patients and greatly improved diagnostic ability.[citation needed]
In 2002, treatment with rapamycin was found to be effective at shrinking tumours in animals. This has led to human trials of rapamycin as a drug to treat several of the tumors associated with TSC.[42]
References
- ↑ 1.0 1.1 1.2 1.3 "Tuberous Sclerosis - Symptoms, Causes, Treatment | NORD". rarediseases.org. Archived from the original on 7 June 2023. Retrieved 9 September 2023.
- ↑ "Tuberous sclerosis - Symptoms and causes". Mayo Clinic. Archived from the original on 9 September 2023. Retrieved 9 September 2023.
- ↑ 3.00 3.01 3.02 3.03 3.04 3.05 3.06 3.07 3.08 3.09 3.10 3.11 3.12 3.13 3.14 3.15 3.16 3.17 3.18 3.19 3.20 3.21 "Tuberous Sclerosis Complex". National Institute of Neurological Disorders and Stroke. Archived from the original on 1 June 2023. Retrieved 9 September 2023.
- ↑ "TUBEROUS SCLEROSIS". www.epilepsydiagnosis.org. Archived from the original on 21 October 2021. Retrieved 9 September 2023.
- ↑ "TUBEROUS SCLEROSIS". www.epilepsydiagnosis.org. Archived from the original on 3 August 2023. Retrieved 9 September 2023.
- ↑ Brigo F, Lattanzi S, Trinka E, Nardone R, Bragazzi NL, Ruggieri M, et al. (December 2018). "First descriptions of tuberous sclerosis by Désiré-Magloire Bourneville (1840-1909)". Neuropathology. 38 (6): 577–582. doi:10.1111/neup.12515. PMID 30215888. S2CID 52269610.
- ↑ 7.0 7.1 Curatolo P, ed. (2003). "Diagnostic Criteria". Tuberous Sclerosis Complex: From Basic Science to Clinical Phenotypes. International review of child neurology. London: Mac Keith Press. ISBN 978-1-898683-39-1. OCLC 53124670.
- ↑ 8.0 8.1 8.2 8.3 8.4 8.5 8.6 8.7 8.8 8.9 Northrup H, Krueger DA (October 2013). "Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference". Pediatric Neurology. 49 (4): 243–54. doi:10.1016/j.pediatrneurol.2013.08.001. PMC 4080684. PMID 24053982.
- ↑ 9.0 9.1 Northrup H, Koenig MK, Pearson DA, Au KS (1993). "Tuberous Sclerosis Complex". In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A (eds.). GeneReviews. Seattle (WA): University of Washington. PMID 20301399. Archived from the original on 27 August 2021. Retrieved 27 January 2019.
- ↑ Ridler K, Suckling J, Higgins N, Bolton P, Bullmore E (September 2004). "Standardized whole brain mapping of tubers and subependymal nodules in tuberous sclerosis complex". Journal of Child Neurology. 19 (9): 658–65. doi:10.1177/08830738040190090501. PMID 15563011. S2CID 23176067.
- ↑ 11.0 11.1 11.2 11.3 11.4 11.5 11.6 de Vries PJ, Wilde L, de Vries MC, Moavero R, Pearson DA, Curatolo P (September 2018). "A clinical update on tuberous sclerosis complex-associated neuropsychiatric disorders (TAND)". American Journal of Medical Genetics. Part C, Seminars in Medical Genetics. 178 (3): 309–320. doi:10.1002/ajmg.c.31637. PMC 6209538. PMID 30117265.
- ↑ Henske EP (December 2003). "Metastasis of benign tumor cells in tuberous sclerosis complex". Genes, Chromosomes & Cancer. 38 (4): 376–81. doi:10.1002/gcc.10252. PMID 14566858. S2CID 22211249.
- ↑ 13.0 13.1 Hinton RB, Prakash A, Romp RL, Krueger DA, Knilans TK (November 2014). "Cardiovascular manifestations of tuberous sclerosis complex and summary of the revised diagnostic criteria and surveillance and management recommendations from the International Tuberous Sclerosis Consensus Group". Journal of the American Heart Association. 3 (6): e001493. doi:10.1161/JAHA.114.001493. PMC 4338742. PMID 25424575.
- ↑ 14.0 14.1 14.2 Randle SC (April 2017). "Tuberous Sclerosis Complex: A Review". Pediatric Annals. 46 (4): e166–e171. doi:10.3928/19382359-20170320-01. PMID 28414398.
- ↑ Arva NC, Pappas JG, Bhatla T, Raetz EA, Macari M, Ginsburg HB, Hajdu CH (January 2012). "Well-differentiated pancreatic neuroendocrine carcinoma in tuberous sclerosis--case report and review of the literature". The American Journal of Surgical Pathology. 36 (1): 149–53. doi:10.1097/PAS.0b013e31823d0560. PMID 22173120.
- ↑ Baraitser M, Patton MA (February 1985). "Reduced penetrance in tuberous sclerosis". Journal of Medical Genetics. 22 (1): 29–31. doi:10.1136/jmg.22.1.29. PMC 1049373. PMID 3981577.
- ↑ van Slegtenhorst M, de Hoogt R, Hermans C, Nellist M, Janssen B, Verhoef S, et al. (August 1997). "Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34". Science. 277 (5327): 805–808. doi:10.1126/science.277.5327.805. PMID 9242607.
- ↑ European Chromosome 16 Tuberous Sclerosis Consortium (December 1993). "Identification and characterization of the tuberous sclerosis gene on chromosome 16". Cell. 75 (7): 1305–15. doi:10.1016/0092-8674(93)90618-Z. PMID 8269512. S2CID 29476292.
- ↑ Brook-Carter PT, Peral B, Ward CJ, Thompson P, Hughes J, Maheshwar MM, Nellist M, Gamble V, Harris PC, Sampson JR (December 1994). "Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease--a contiguous gene syndrome". Nature Genetics. 8 (4): 328–32. doi:10.1038/ng1294-328. PMID 7894481. S2CID 23793670.
- ↑ Dabora SL, Jozwiak S, Franz DN, Roberts PS, Nieto A, Chung J, Choy YS, Reeve MP, Thiele E, Egelhoff JC, Kasprzyk-Obara J, Domanska-Pakiela D, Kwiatkowski DJ (January 2001). "Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs". American Journal of Human Genetics. 68 (1): 64–80. doi:10.1086/316951. PMC 1234935. PMID 11112665.
- ↑ Rendtorff ND, Bjerregaard B, Frödin M, Kjaergaard S, Hove H, Skovby F, Brøndum-Nielsen K, Schwartz M (October 2005). "Analysis of 65 tuberous sclerosis complex (TSC) patients by TSC2 DGGE, TSC1/TSC2 MLPA, and TSC1 long-range PCR sequencing, and report of 28 novel mutations". Human Mutation. 26 (4): 374–83. doi:10.1002/humu.20227. PMID 16114042. S2CID 10571695.
- ↑ Pal R, Xiong Y, Sardiello M (February 2019). "Abnormal glycogen storage in tuberous sclerosis complex caused by impairment of mTORC1-dependent and -independent signaling pathways". Proc Natl Acad Sci U S A. 116 (8): 2977–2986. doi:10.1073/pnas.1812943116. PMC 6386676. PMID 30728291.
- ↑ Crino PB, Nathanson KL, Henske EP (September 2006). "The tuberous sclerosis complex". The New England Journal of Medicine. 355 (13): 1345–56. CiteSeerX 10.1.1.319.8438. doi:10.1056/NEJMra055323. PMID 17005952.
- ↑ "Tuberous Sclerosis Complex". University Hospitals Birmingham NHS Foundation Trust. Archived from the original on 16 December 2018. Retrieved 16 December 2018.
- ↑ 25.0 25.1 25.2 Krueger DA, Northrup H (October 2013). "Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference". Pediatric Neurology. 49 (4): 255–65. doi:10.1016/j.pediatrneurol.2013.08.002. PMC 4058297. PMID 24053983.
- ↑ "Press Announcements - FDA approves Afinitor for non-cancerous kidney tumors caused by rare genetic disease". www.fda.gov. Archived from the original on 11 February 2017. Retrieved 2017-02-08.
- ↑ "FDA Approval for Everolimus". National Cancer Institute. 2009-04-21. Archived from the original on 11 February 2017. Retrieved 2017-02-08.
- ↑ Sasongko TH, Ismail NF, Zabidi-Hussin Z (July 2016). "Rapamycin and rapalogs for tuberous sclerosis complex". The Cochrane Database of Systematic Reviews. 7: CD011272. doi:10.1002/14651858.CD011272.pub2. PMC 6458010. PMID 27409709.
- ↑ French JA, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, Curatolo P, de Vries PJ, Dlugos DJ, Berkowitz N, Voi M, Peyrard S, Pelov D, Franz DN (October 2016). "Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study". Lancet. 388 (10056): 2153–63. doi:10.1016/s0140-6736(16)31419-2. PMID 27613521.
- ↑ Capal JK, Franz DN (2016). "Profile of everolimus in the treatment of tuberous sclerosis complex: an evidence-based review of its place in therapy". Neuropsychiatric Disease and Treatment. 12: 2165–72. doi:10.2147/NDT.S91248. PMC 5003595. PMID 27601910.
- ↑ Novartis International AG. "Novartis drug Votubia® receives EU approval to treat refractory partial-onset seizures in patients with TSC". GlobeNewswire News Room. Archived from the original on 11 February 2017. Retrieved 2017-02-08.
- ↑ Asano E, Juhász C, Shah A, Muzik O, Chugani DC, Shah J, Sood S, Chugani HT (July 2005). "Origin and propagation of epileptic spasms delineated on electrocorticography". Epilepsia. 46 (7): 1086–97. doi:10.1111/j.1528-1167.2005.05205.x. PMC 1360692. PMID 16026561.
- ↑ Chugani HT, Luat AF, Kumar A, Govindan R, Pawlik K, Asano E (August 2013). "α-[11C]-Methyl-L-tryptophan--PET in 191 patients with tuberous sclerosis complex". Neurology. 81 (7): 674–80. doi:10.1212/WNL.0b013e3182a08f3f. PMC 3775695. PMID 23851963.
- ↑ Hong AM, Turner Z, Hamdy RF, Kossoff EH (August 2010). "Infantile spasms treated with the ketogenic diet: prospective single-center experience in 104 consecutive infants". Epilepsia. 51 (8): 1403–407. doi:10.1111/j.1528-1167.2010.02586.x. PMID 20477843. S2CID 26666421.
- ↑ Jacks SK, Witman PM (September–October 2015). "Tuberous Sclerosis Complex: An Update for Dermatologists". Pediatric Dermatology. 32 (5): 563–70. doi:10.1111/pde.12567. PMID 25776100. S2CID 72874.
- ↑ "Tuberous Sclerosis Fact Sheet". National Institute of Neurological Disorders and Stroke. 2018-07-06. Archived from the original on 16 December 2018. Retrieved 16 December 2018.
- ↑ Samir H, Ghaffar HA, Nasr M (March 2011). "Seizures and intellectual outcome: clinico-radiological study of 30 Egyptian cases of tuberous sclerosis complex". European Journal of Paediatric Neurology. 15 (2): 131–37. doi:10.1016/j.ejpn.2010.07.010. PMID 20817577.
- ↑ Shepherd CW, Gomez MR, Lie JT, Crowson CS (August 1991). "Causes of death in patients with tuberous sclerosis". Mayo Clinic Proceedings. 66 (8): 792–96. doi:10.1016/s0025-6196(12)61196-3. PMID 1861550.
- ↑ Rakowski SK, Winterkorn EB, Paul E, Steele DJ, Halpern EF, Thiele EA (November 2006). "Renal manifestations of tuberous sclerosis complex: Incidence, prognosis, and predictive factors". Kidney International. 70 (10): 1777–82. doi:10.1038/sj.ki.5001853. PMID 17003820.
- ↑ O'Callaghan FJ, Shiell AW, Osborne JP, Martyn CN (May 1998). "Prevalence of tuberous sclerosis estimated by capture-recapture analysis". Lancet. 351 (9114): 1490. doi:10.1016/S0140-6736(05)78872-3. PMID 9605811. S2CID 9262685.
- ↑ Curatolo P, ed. (2003). "Historical Background". Tuberous Sclerosis Complex: From Basic Science to Clinical Phenotypes. International review of child neurology. London: Mac Keith Press. ISBN 978-1-898683-39-1. OCLC 53124670.
- ↑ Rott HD, Mayer K, Walther B, Wienecke R (March 2005). "Zur Geschichte der Tuberösen Sklerose (The History of Tuberous Sclerosis)" (PDF) (in Deutsch). Tuberöse Sklerose Deutschland e.V. Archived from the original (PDF) on 15 March 2007. Retrieved 8 January 2007.
External links
| Classification | |
|---|---|
| External resources |
- tuberous-sclerosis at NIH/UW GeneTests
- GeneReview/NCBI/NIH/UW entry on Tuberous Sclerosis Complex Archived 27 August 2021 at the Wayback Machine
- Pages with script errors
- CS1 Deutsch-language sources (de)
- All articles with unsourced statements
- Articles with unsourced statements from December 2020
- Articles with invalid date parameter in template
- Articles with unsourced statements from September 2020
- Articles with hatnote templates targeting a nonexistent page
- Webarchive template wayback links
- Autosomal dominant disorders
- Genodermatoses
- Rare diseases
- Biology of attention deficit hyperactivity disorder
- Autism
- Intellectual disability
- Biology of obsessive–compulsive disorder
- Disorders causing seizures
- RTT
- RTTILAE