Tertiary hyperparathyroidism
| Tertiary Hyperparathyroidism | |
|---|---|
  | |
| |
| Specialty | Endocrinology |
| Symptoms | None, kidney stones, weakness, depression, bone pains, confusion, increased urination |
| Complications | Osteoporosis |
| Usual onset | 50 to 60 |
| Types | Primary, secondary, tertiary |
| Causes | Tertiary: parathyroid adenoma, multiple benign tumors, parathyroid cancer, parathyroid hyperplasia, growth of parathyroid tissue, secondary hyperparathyroidism |
| Diagnostic method | High blood calcium and high PTH levels |
| Treatment | Surgery, intravenous normal saline |
| Frequency | ~2 per 1,000 |
Tertiary hyperparathyroidism is a condition involving the overproduction of the hormone, parathyroid hormone, produced by the parathyroid glands.[1] The parathyroid glands are involved in monitoring and regulating blood calcium levels and respond by either producing or ceasing to produce parathyroid hormone. Anatomically, these glands are located in the neck, para-lateral to the thyroid gland, which does not have any influence in the production of parathyroid hormone. Parathyroid hormone is released by the parathyroid glands in response to low blood calcium circulation. Persistent low levels of circulating calcium are thought to be the catalyst in the progressive development of adenoma, in the parathyroid glands resulting in primary hyperparathyroidism. While primary hyperparathyroidism is the most common form of this condition,[2][3][4] secondary and tertiary are thought to result due to chronic kidney disease (CKD).[2] Estimates of CKD prevalence in the global community range from 11 to 13% which translate to a large portion of the global population at risk of developing tertiary hyperparathyroidism.[5] Tertiary hyperparathyroidism was first described in the late 1960s and had been misdiagnosed as primary prior to this.[6] Unlike primary hyperparathyroidism, the tertiary form presents as a progressive stage of resolved secondary hyperparathyroidism with biochemical hallmarks that include elevated calcium ion levels in the blood, hypercalcemia, along with autonomous production of parathyroid hormone and adenoma in all four parathyroid glands.[1] Upon diagnosis treatment of tertiary hyperparathyroidism usually leads to a surgical intervention.[7]
Signs and symptoms
Symptoms in tertiary hyperparathyroidism are generally those seen in relation to elevated blood calcium levels.[8][1] Tertiary hyperparathyroidism shares many symptomatic features with that of primary hyperparathyroidism, as the two are defined by hypercalcemia. These symptoms can vary greatly from asymptomatic to conditions leading to decreased quality of life.[1][4]
Non-specific symptoms include feeling tired and thirsty, mood changes including, feeling blue, weak and irritable along with other symptoms like itching, headache, joint pain, forgetfulness and abdominal pain have also been noted.[8][4][9][1] More specific symptoms related to elevated blood calcium and phosphate levels include bone pain or osteodynia and tenderness which are common and related to proximal muscle tenderness. Other signs can include pancreatitis, kidney stones, corneal calcifications, thinning of long bones, and hypodermic calcifications which may be palpable in some patients.[2][4][1]
Calciphylaxis, though uncommon, can develop in patients with tertiary hyperparathyroidism. The product of elevated calcium and phosphate, forming crystal structures, that are then deposited in blood vessels. These crystals cause an inflammatory response and can lead to the occlusion of smaller vessels. Further complications like secondary infections and necrosis can develop from this and can be fatal for some, making the monitoring of blood calcium and phosphate levels necessary.[8][1]
Conditions due to bone loss such as osteopenia and osteoporosis are common in tertiary hyperparathyroidism along with pathologic fractures. Pseudoclubbing of the digits can also be indicative of a severe tertiary hyperparathyroidism due to excess resorption at the distal phalanges.[8][1]
Diagnosis includes both clinical and laboratory investigations. Radiological investigations include looking for signs of bone loss in both the hands and pelvis which is characteristic of tertiary hyperparathyroidism.[8] Other clinical examination can include grading of muscle weakness, which is done by asking the patient to stand from a seated position with their hands folded across their chest.[4][8] Laboratory investigations include evaluating blood calcium and alkaline phosphatase, which are always increased in tertiary hyperparathyroidism. Other common results from laboratory investigations would include decreased vitamin D levels, elevated blood parathyroid hormone and hyperphosphatemia.[9][8][1][4]
Cause


Hyperparathyroidism, in general, is caused by either tumorous growth in one or more parathyroid glands or a prolonged decrease in blood calcium levels or hypocalcaemia which in turn stimulates the production of parathyroid hormone release from the parathyroid gland.[10][11] The parathyroid gland is located beside the thyroid gland in the neck, below and in front of the larynx and above the trachea. It is composed of four glands in total that monitor blood calcium levels via the calcium sensing receptors, a g-coupled protein receptor.[12] The parathyroid glands main role is calcium homeostasis.[13][12] Histologically, these glands are composed of chief cells and oxyphil cells with the chief cell primarily responsible for the storing and release of parathyroid hormone. These cells are arranged in a pseudo-follicular pattern similar to the thyroid follicles. Keratin staining is used to image the parathyroid hormone granules.[9][14]
Parathyroid hormone is responsible for the induction of increased calcium absorption in the gastrointestinal tract or gut and in the kidney. It also induces calcium and phosphate resorption from the bone by osteoclasts.[15][11] Parathyroid hormone also plays a role in activating vitamin D from its pro form to its active form.[15] Vitamin D is also responsible for increased blood calcium levels and works in conjunction with parathyroid hormone. Vitamin D is also partly responsible for the inhibition of parathyroid hormone release by binding Vitamin D receptors at the parathyroid gland.[11]
Tertiary hyperparathyroidism is defined by autonomous release of parathyroid hormone while in a hypercalcaemic state. Unlike primary hyperparathyroidism, hypercalcemia in the tertiary form is thought to be the result of resolution of secondary hyperparathyroidism rather than adenoma formation alone.[4][11][10]
Many of the mechanisms that drive the formation of tertiary hyperparathyroidism are due to outcomes of secondary hyperparathyroidism and so the tertiary from is said to be a continued progressive hyperparathyroidism.[10][11] Secondary hyperparathyroidism occurs mainly in those who have chronic kidney disease or vitamin D deficiencies both of which lead to malabsorption of calcium and phosphate leading to decreased blood calcium levels inducing a hyperparathyroidism. Hyperphosphatemia in secondary hyperparathyroidism, due to increased parathyroid hormone, is thought to act directly on parathyroid glands and induce a hyperplasia or increased growth of the chief cells in particular.[11] At the same time the hyperplasic parathyroid glands have reduced fibroblast-growth-factor-23 (FGF-23) and vitamin D receptor expression. FGF-23 is partly responsible for phosphate homeostasis and provides negative feedback to the parathyroid gland as does vitamin D.[16][17][11]
During prolonged secondary hyperparathyroidism increased blood phosphate levels drive hyperplasia of the parathyroid gland and this acts to reset calcium sensitivity at the calcium sensing receptors leading to tertiary hyperparathyroidism after resolution of the secondary form with the continued release of parathyroid hormone in the presence of hypercalcemia.[11]
Risk Factors
An elevated risk of developing tertiary hyperparathyroidism exists when late stage kidney disease is not corrected timely.[7][4] This is due to a hyperphosphatemia acting directly on the parathyroid glands. Genetically, those who have an X-linked dominant disorder that disrupts phosphate transport at the renal tubules (X-Linked hypophosphatemic rickets) and are receiving oral phosphate treatment have shown to be at high risk of developing tertiary hyperparathyroidism in the absence of secondary hyperparathyroidism.[18] Recurring tertiary hyperparathyroidism is generally seen to be caused by incomplete parathyroidectomy without renal transplant and the risk is increased when the parathyroid tissue left after surgery is that of a nodular type.[7]
Other risk factors of tertiary hyperparathyroidism include an elevated risk of developing acute pancreatitis, mainly due to the hypercalcemia associated with the hyperparathyroidism.[19] Other studies have shown a significant increase in the risk of developing malignancies of the urinary tract and renal system with women being more at risk.[20] Though there is some conjecture as to the correlation between hyperparathyroidism and thyroid carcinoma development, there is however a correlation between the two, which is thought to be due to prolonged irradiation of the neck and head for parathyroid adenomas and increased parathyroid hormone.[21]
Other studies have found some correlation in the development of renal disease following parathyroidectomy. However, the mechanism for this effect remains unknown.[22]
Pathophysiology
-
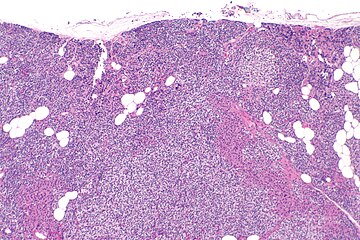
Parathyroid hyperplasia low mag.
-

Parathyroid hyperplasia medium mag.
-
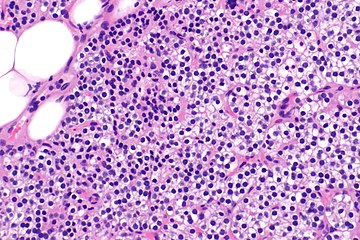
Parathyroid hyperplasia high mag.



Tertiary Hyperparathyroidism is almost always related to end stage kidney disease and a secondary hyperparathyroidism.[23][4][8] Physiological changes due to the kidney damage adversely affect feedback loops that control secretion of parathyroid hormone. Renal management of phosphate is impaired in secondary hyperparathyroidism which results in hyperphosphatemia.[4][6]
Primary hyperplasia of the parathyroid gland, results from both hypocalcaemia and increased phosphate levels by decreasing expression of calcium sensing receptors and vitamin D receptors at the parathyroid gland.[8][4] These decreases in receptor expression lead to hyperfunctioning of the parathyroid. Hyperfunction of the parathyroid gland is thought to exacerbate primary hyperplasia which evolves further to a secondary more aggressive hyperplasia. Histologically, these hyperplasic glands can be either diffuse or nodular.[24] Primary hyperplasia, usually resulting in diffuse polyclonal growth is manly related to reversible secondary hyperparathyroidism. Secondary hyperplasia of the parathyroid gland is more often a nodular, monoclonal growth that sustains secondary hyperparathyroidism and is the catalyst in the progression to tertiary hyperparathyroidism. Nodular hyperplastic glands in tertiary hyperparathyroidism are distinctly larger in both absolute size and weight up to 20-40-fold increases have been reported.[25][26][24]
Parathyroid glands are normally composed of chief cells, adipocytes and scattered oxyphil cells.[27][14] Chief cells are thought to be responsible for the production, storage and secretion of parathyroid hormone. These cells appear light and dark with a prominent Golgi body and endoplasmic reticulum. In electron micrographs, secretory vesicles can be seen in and around the Golgi and at the cell membrane. These cells also contain prominent cytoplasmic adipose.[27][14] Upon onset of hyperplasia these cells are described as having a nodular pattern with enlargement of protein synthesis machinery such as the endoplasmic reticulum and Golgi. Increased secretory vesicles are seen and decreased intercellular fat is characteristic.[27][24] Oxyphil cells also appear hyperplasic however, these cells are much less prominent.[citation needed]
Biochemically, there are changes in function between normal and nodular hyperplastic parathyroid glands. These changes involve proto-oncogene expression and activation of proliferative pathways while inactivating apoptotic pathways.[28] In nodular parathyroid tissue increased expression of TGF-a, a growth factor, and EGFR, its receptor, results in aggressive proliferation and further downregulation of vitamin D receptors, which act to suppress hormone secretions.[25][8][28] Furthermore, the proliferative marker, Ki67 is seen to be highly expressed in the secondary nodular hyperplastic state.[28][25] Tumour suppressor genes have also been highlighted as being silenced or degraded in nodular hyperplastic parathyroid tissue.[8][28] One such gene, p53, has been shown to regulate multiple tumour suppressor pathways and in tumorigenesis can be degraded by b-catenin. This pathway, in some aspect, is mediated by CACYBP, which is highly expressed in nodular parathyroid hyperplasia.[28]
Diagnosis
In terms of the diagnosis of Tertiary hyperparathyroidism we find that both high blood calcium and high PTH levels are indicative of this condition[29]
Treatment

Early pharmaceutical treatment for tertiary hyperparathyroidism may include supplementing vitamin D and the use of cinacalcet.[4][1][30] Cinacalcet acts to increase the sensitivity of the calcium sensing receptors to calcium leading to a reduction in parathyroid hormone release, however, its use has limited impact in those with tertiary hyperparathyroidism.[30] These treatments are more likely only transient therapies before parathyroidectomy is performed. Indications for surgery in tertiary hyperparathyroidism commonly involve the development of chronic, severe conditions including osteopenia, persistent severe hypercalcemia, bone pain and pathologic fracture.[7][2][1][4] Other indications include development of conditions such calciphylaxis.[1] Surgical options for tertiary hyperparathyroidism include subtotal parathyroidectomy (three and one half of total tissue) and total parathyroidectomy with autotransplatation of resected tissue.[7][2][1] Outcomes from surgery are generally favourable and a return to normalised blood calcium levels and parathyroid function is seen.[1]
History
In 1962, Dr C.E Dent reported that autonomous hyperparathyroidism may result from malabsorption syndromes and chronic kidney disease.[6] The term 'tertiary hyperparathyroidism' was first used in 1963 by Dr Walter St. Gaur to describe a case reported on at Massachusetts General hospital.[6] This case involved a patient who had presented with autonomous parathyroid adenoma causing hypercalcemia with a background of parathyroid hyperplasia. Further reports were recorded in 1964, 65 and 67 of suspected tertiary hyperparathyroidism.[citation needed]
In 1968 Davies, Dent and Watson produced a historic case study where they reviewed 200 cases of previously diagnosed primary hyperparathyroidism and found the majority of these cases should be reclassified as tertiary.[6] These were important findings as it allowed an understanding into distinguishing features of primary, secondary and tertiary hyperparathyroidism which then allows appropriate medical treatment.[citation needed]
It is now understood that tertiary hyperparathyroidism is defined as the presence of hypercalcemia, hyperphosphatemia and parathyroid hormone due to terminally biased parathyroid-bone-kidney feedback loop.[4] Although there is still conjecture as to whether tertiary hyperparathyroidism is also due to adenomatous growth or hyperplasia it is clear that tertiary hyperparathyroidism presents with some form of tissue enlargement in all four parathyroid glands.[7][24]
See also
References
- ↑ 1.00 1.01 1.02 1.03 1.04 1.05 1.06 1.07 1.08 1.09 1.10 1.11 1.12 1.13 Pitt SC, Sippel RS, Chen H (October 2009). "Secondary and tertiary hyperparathyroidism, state of the art surgical management". The Surgical Clinics of North America. 89 (5): 1227–39. doi:10.1016/j.suc.2009.06.011. PMC 2905047. PMID 19836494.
- ↑ 2.0 2.1 2.2 2.3 2.4 Callender GG, Carling T, Christison-Lagay E, Udelsman R (2016). "Chapter 65 - Surgical Management of Hyperparathyroidism". In Jameson JL, De Groot LJ, de Kretser DM, Giudice LC (eds.). Endocrinology: Adult and Pediatric (Seventh ed.). W.B. Saunders. pp. 1135–1146.e3. doi:10.1016/b978-0-323-18907-1.00065-2. ISBN 978-0-323-18907-1.
- ↑ Clayman GL, Gonzalez HE, El-Naggar A, Vassilopoulou-Sellin R (March 2004). "Parathyroid carcinoma: evaluation and interdisciplinary management". Cancer (Seventh ed.). W.B. Saunders. 100 (5): 900–5. doi:10.1016/b978-0-323-18907-1.00065-2. ISBN 978-0-323-18907-1. PMID 14983483.
- ↑ 4.00 4.01 4.02 4.03 4.04 4.05 4.06 4.07 4.08 4.09 4.10 4.11 4.12 4.13 van der Plas WY, Noltes ME, van Ginhoven TM, Kruijff S (July 2019). "Secondary and Tertiary Hyperparathyroidism: A Narrative Review". Scandinavian Journal of Surgery. 109 (4): 271–278. doi:10.1177/1457496919866015. PMID 31364494.
- ↑ Hill NR, Fatoba ST, Oke JL, Hirst JA, O'Callaghan CA, Lasserson DS, Hobbs FD (2016-07-06). Remuzzi G (ed.). "Global Prevalence of Chronic Kidney Disease - A Systematic Review and Meta-Analysis". PLOS ONE. 11 (7): e0158765. Bibcode:2016PLoSO..1158765H. doi:10.1371/journal.pone.0158765. PMC 4934905. PMID 27383068.
- ↑ 6.0 6.1 6.2 6.3 6.4 Davies DR, Dent CE, Watson L (August 1968). "Tertiary hyperparathyroidism". British Medical Journal. 3 (5615): 395–9. doi:10.1136/bmj.3.5615.395. PMC 1986316. PMID 5691200.
- ↑ 7.0 7.1 7.2 7.3 7.4 7.5 Gasparri G, Camandona M, Abbona GC, Papotti M, Jeantet A, Radice E, et al. (January 2001). "Secondary and tertiary hyperparathyroidism: causes of recurrent disease after 446 parathyroidectomies". Annals of Surgery. 233 (1): 65–9. doi:10.1097/00000658-200101000-00011. PMC 1421168. PMID 11141227.
- ↑ 8.00 8.01 8.02 8.03 8.04 8.05 8.06 8.07 8.08 8.09 8.10 Khan AA, Clark OH (2012). Handbook of parathyroid diseases : a case-based practical guide. New York: Springer. ISBN 978-1-4614-2164-1. OCLC 778874657.
- ↑ 9.0 9.1 9.2 Roychowdhury M (2013). "Anatomy and histology". www.pathologyoutlines.com. Archived from the original on 2020-10-22. Retrieved 2020-04-22.
- ↑ 10.0 10.1 10.2 Jamal SA, Miller PD (January 2013). "Secondary and tertiary hyperparathyroidism". Journal of Clinical Densitometry. 16 (1): 64–8. doi:10.1016/j.jocd.2012.11.012. PMID 23267748.
- ↑ 11.0 11.1 11.2 11.3 11.4 11.5 11.6 11.7 Kennel KA, Clarke BL (2016). "Tertiary Hyperparathyroidism". In Kearns AE, Wermers RA (eds.). Hyperparathyroidism. Springer International Publishing. pp. 179–188. doi:10.1007/978-3-319-25880-5_20. ISBN 978-3-319-25878-2.
- ↑ 12.0 12.1 Hendy GN, Goltzman D (July 2011). Parathyroid anatomy, hormone synthesis, secretion, action, and receptors. Oxford Textbook of Endocrinology and Diabetes. Oxford University Press. pp. 633–641. doi:10.1093/med/9780199235292.003.0404. ISBN 978-0-19-923529-2.
- ↑ Goltzman D (December 2018). "Physiology of Parathyroid Hormone". Endocrinology and Metabolism Clinics of North America. 47 (4): 743–758. doi:10.1016/j.ecl.2018.07.003. PMID 30390810. S2CID 53216606.
- ↑ 14.0 14.1 14.2 Brown MB, Limaiem F (2020). Histology, Parathyroid Gland. StatPearls. StatPearls Publishing. PMID 31536203. Archived from the original on 2021-03-03. Retrieved 2020-04-22.
- ↑ 15.0 15.1 Goltzman D, Mannstadt M, Marcocci C (2018). Giustina A, Bilezikian JP (eds.). "Physiology of the Calcium-Parathyroid Hormone-Vitamin D Axis". Frontiers of Hormone Research. S. Karger AG. 50: 1–13. doi:10.1159/000486060. ISBN 978-3-318-06338-7. PMID 29597231.
- ↑ Silver J, Naveh-Many T (2012). Kuro-o M (ed.). "FGF23 and the parathyroid". Advances in Experimental Medicine and Biology. Springer US. 728: 92–9. doi:10.1007/978-1-4614-0887-1_6. ISBN 978-1-4614-0886-4. PMID 22396164.
- ↑ Komaba H, Fukagawa M (February 2010). "FGF23-parathyroid interaction: implications in chronic kidney disease". Kidney International. 77 (4): 292–8. doi:10.1038/ki.2009.466. PMID 20010546.
- ↑ Mäkitie O, Kooh SW, Sochett E (February 2003). "Prolonged high-dose phosphate treatment: a risk factor for tertiary hyperparathyroidism in X-linked hypophosphatemic rickets". Clinical Endocrinology. 58 (2): 163–8. doi:10.1046/j.1365-2265.2003.01685.x. PMID 12580931. S2CID 20053408.
- ↑ Stephani J, Akinli AS, von Figura G, Barth TF, Weber T, Hartmann B, et al. (September 2011). "Acute Pancreatitis in a patient with hypercalcemia due to tertiary hyperparathyroidism". Zeitschrift für Gastroenterologie (in Deutsch). 49 (9): 1263–6. doi:10.1055/s-0029-1245981. PMID 21887663.
- ↑ Pickard AL, Gridley G, Mellemkjae L, Johansen C, Kofoed-Enevoldsen A, Cantor KP, Brinton LA (October 2002). "Hyperparathyroidism and subsequent cancer risk in Denmark". Cancer. 95 (8): 1611–7. doi:10.1002/cncr.10846. PMID 12365007. S2CID 35173002.
- ↑ Burmeister LA, Sandberg M, Carty SE, Watson CG (April 1997). "Thyroid carcinoma found at parathyroidectomy: association with primary, secondary, and tertiary hyperparathyroidism". Cancer. 79 (8): 1611–6. doi:10.1002/(SICI)1097-0142(19970415)79:8<1611::AID-CNCR26>3.0.CO;2-#. PMID 9118047. S2CID 196356726.
- ↑ Montenegro FL, Martin RM, Corrêa PH (April 2009). "Renal failure after surgery for primary hyperparathyroidism: is acute reduction of parathyroid function a risk factor?". Clinics. 64 (4): 369–72. doi:10.1590/S1807-59322009000400017. PMC 2694470. PMID 19488597.
- ↑ Tai TS, Hsu YH, Chang JM, Chen CC (January 2019). "Recurrent tertiary hyperparathyroidism due to supernumerary parathyroid glands in a patient receiving long-term hemodialysis: a case report". BMC Endocrine Disorders. 19 (1): 16. doi:10.1186/s12902-019-0346-7. PMC 6350335. PMID 30691427.
- ↑ 24.0 24.1 24.2 24.3 Krause MW, Hedinger CE (August 1985). "Pathologic study of parathyroid glands in tertiary hyperparathyroidism". Human Pathology. 16 (8): 772–84. doi:10.1016/S0046-8177(85)80248-3. PMID 4018775.
- ↑ 25.0 25.1 25.2 Vulpio C, Bossola M, Di Stasio E, Tazza L, Silvestri P, Fadda G (June 2013). "Histology and immunohistochemistry of the parathyroid glands in renal secondary hyperparathyroidism refractory to vitamin D or cinacalcet therapy". European Journal of Endocrinology. 168 (6): 811–9. doi:10.1530/EJE-12-0947. PMID 23520248. Archived from the original on 2023-03-04. Retrieved 2022-09-17.
- ↑ Lomonte C, Vernaglione L, Chimienti D, Bruno A, Cocola S, Teutonico A, et al. (May 2008). "Does vitamin D receptor and calcium receptor activation therapy play a role in the histopathologic alterations of parathyroid glands in refractory uremic hyperparathyroidism?". Clinical Journal of the American Society of Nephrology. 3 (3): 794–9. doi:10.2215/CJN.04150907. PMC 2386693. PMID 18322048.
- ↑ 27.0 27.1 27.2 Altmann HW, Benirschke K, Bohle A, Brinkhous KM, Cohrs P, Cottier H, Eder M, Gedigk P, Giese W, eds. (1972). Current Topics in Pathology / Ergebnisse der Pathologie. Berlin, Heidelberg: Springer Berlin Heidelberg. doi:10.1007/978-3-642-65324-7. ISBN 978-3-642-65326-1. S2CID 44281698.
- ↑ 28.0 28.1 28.2 28.3 28.4 Týcová I, Sulková SD, Štěpánková J, Krejčík Z, Merkerová MD, Stránecký V, et al. (October 2016). "Molecular patterns of diffuse and nodular parathyroid hyperplasia in long-term hemodialysis". American Journal of Physiology. Endocrinology and Metabolism. 311 (4): E720–E729. doi:10.1152/ajpendo.00517.2015. PMID 27600827.
- ↑ Deftos, L. J. (1 January 2001). "CHAPTER 9 - Immunoassays for PTH and PTHrP: Clinical Applications". The Parathyroids (Second Edition). Academic Press. pp. 143–165. ISBN 978-0-12-098651-4. Retrieved 4 March 2023.
- ↑ 30.0 30.1 Dulfer RR, Koh EY, van der Plas WY, Engelsman AF, van Dijkum EJ, Pol RA, et al. (February 2019). "Parathyroidectomy versus cinacalcet for tertiary hyperparathyroidism; a retrospective analysis". Langenbeck's Archives of Surgery. 404 (1): 71–79. doi:10.1007/s00423-019-01755-4. PMC 6394681. PMID 30729318.
External links
| Classification |
|---|
- Pages using duplicate arguments in template calls
- Pages with script errors
- CS1 Deutsch-language sources (de)
- All articles with unsourced statements
- Articles with unsourced statements from October 2021
- Articles with invalid date parameter in template
- Articles with hatnote templates targeting a nonexistent page
- Parathyroid disorders