Sturge–Weber syndrome
| Sturge–Weber syndrome | |
|---|---|
| Other names: Sturge–Weber–Krabbe disease | |
 | |
| CT scan of Sturge-Weber syndrome | |
Sturge–Weber syndrome, sometimes referred to as encephalotrigeminal angiomatosis, is a rare congenital neurological and skin disorder. It is one of the phakomatoses and is often associated with port-wine stains of the face, glaucoma, seizures, intellectual disability, and ipsilateral leptomeningeal angioma (cerebral malformations and tumors). Sturge–Weber syndrome can be classified into three different types. Type 1 includes facial and leptomeningeal angiomas as well as the possibility of glaucoma or choroidal lesions. Normally, only one side of the brain is affected. This type is the most common. Type 2 involvement includes a facial angioma (port wine stain) with a possibility of glaucoma developing. There is no evidence of brain involvement. Symptoms can show at any time beyond the initial diagnosis of the facial angioma. The symptoms can include glaucoma, cerebral blood flow abnormalities and headaches. More research is needed on this type of Sturge–Weber syndrome. Type 3 has leptomeningeal angioma involvement exclusively. The facial angioma is absent and glaucoma rarely occurs. This type is only diagnosed via brain scan.[1]
Sturge–Weber is an embryonal developmental anomaly resulting from errors in mesodermal and ectodermal development. Unlike other neurocutaneous disorders (phakomatoses), Sturge–Weber occurs sporadically (i.e., does not have a hereditary cause). It is caused by a mosaic, somatic activating mutation occurring in the GNAQ gene.[2] Imaging findings may include tram track calcifications on CT, pial angiomatosis, and hemicerebral atrophy.[3]
Signs and symptoms
Sturge–Weber syndrome is usually manifested at birth by a port-wine stain on the forehead and upper eyelid of one side of the face, or the whole face. The birthmark can vary in color from light pink to deep purple and is caused by an overabundance of capillaries around the ophthalmic branch of the trigeminal nerve, just under the surface of the face. There is also malformation of blood vessels in the pia mater overlying the brain on the same side of the head as the birthmark. This causes calcification of tissue and loss of nerve cells in the cerebral cortex.[citation needed]
Neurological signs include seizures that begin in infancy and may worsen with age. Convulsions usually happen on the side of the body opposite the birthmark, and vary in severity. There may also be muscle weakness on the side of the body opposite the birthmark.[citation needed]
Some children will have developmental delays and cognitive delays; about 50% will have glaucoma (optic neuropathy often associated with increased intraocular pressure), which can be present at birth or develop later. Glaucoma can be expressed as leukocoria, which should suggest further evaluation for retinoblastoma. Increased pressure within the eye can cause the eyeball to enlarge and bulge out of its socket (buphthalmos).[citation needed]
-
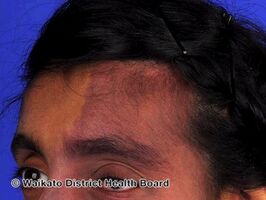
Sturge-Weber syndrome
-
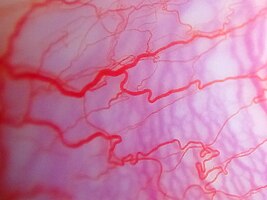
Dilated bulbar vessels in Sturge–Weber syndrome
.jpg)

Sturge–Weber syndrome rarely affects other body organs.[citation needed]
| Presentation[4] | |
|---|---|
| Seizures | 75–90% |
| Vascular headache | 40–60% |
| Developmental (cognitive) delay | 50–70% |
| Glaucoma | 30–70% |
| Hemianopsia | 40–45% |
| Hemiparesis | 25–60% |
Cause
The blood vessel formations associated with SWS start in the fetal stage. Around the sixth week of development, a network of nerves develops around the area that will become a baby's head. Normally, this network goes away in the ninth week of development. In babies with SWS due to mutation of gene GNAQ, this network of nerves doesn't go away. This reduces the amount of oxygen and blood flowing to the brain, which can affect brain tissue development.[citation needed]
Diagnosis
CT and MRI are most often used to identify intracranial abnormalities. When a child is born with a facial cutaneous vascular malformation covering a portion of the upper or the lower eyelids, imaging should be performed to screen for intracranial leptomeningeal angiomatosis. The haemangioma present on the surface of the brain is in the vast majority of cases on the same side as the birth mark and gradually results in calcification of the underlying brain and atrophy of the affected region.[5]
Treatment
Treatment for Sturge–Weber syndrome is symptomatic. Laser treatment may be used to lighten or remove the birthmark. Anticonvulsant medications may be used to control seizures. Doctors recommend early monitoring for glaucoma, and surgery may be performed on more serious cases. When one side of the brain is affected and anticonvulsants prove ineffective, the standard treatment is neurosurgery to remove or disconnect the affected part of the brain (hemispherectomy). Physical therapy should be considered for infants and children with muscle weakness. Educational therapy is often prescribed for those with intellectual disability or developmental delays, but there is no complete treatment for the delays. Brain surgery involving removing the portion of the brain that is affected by the disorder can be successful in controlling the seizures so that the patient has only a few seizures that are much less intense than pre-surgery. Surgeons may also opt to "switch-off" the affected side of the brain.[6]
Latanoprost (Xalatan), a prostaglandin, may significantly reduce IOP (intraocular pressure) in patients with glaucoma associated with Sturge–Weber syndrome. Latanoprost is commercially formulated as an aqueous solution in a concentration of 0.005% preserved with 0.02% benzalkonium chloride (BAC). The recommended dosage of latanoprost is one drop daily in the evening, which permits better diurnal IOP control than does morning instillation. Its effect is independent of ethnicity, gender or age, and it has few to no side effects. Contraindications include a history of cystic macular edema (CME), epiretinal membrane formation, vitreous loss during cataract surgery, history of macular edema associated with branch retinal vein occlusion, history of anterior uveitis, and diabetes mellitus. It is also wise to advise patients that unilateral treatment can result in heterochromia or hypertrichosis that may become cosmetically objectionable.[citation needed]
| Complication[4] | Treatment (1st choice) | Treatment (2nd choice) |
|---|---|---|
| Glaucoma | Beta blocker drops | Adrenergic drops |
| Partial Epilepsy | Carbamazepine | Valproate, Topiramate |
| Headache | Ibuprofen | Sumatriptan |
| Strokelike Episodes | Aspirin | None |
| Neurobehavior | Methylphenidate | Clonidine |
Prognosis
Although it is possible for the birthmark and atrophy in the cerebral cortex to be present without symptoms, most infants will develop convulsive seizures during their first year of life. There is a greater likelihood of intellectual impairment when seizures are resistant to treatment. Studies do not support the widely held belief that seizure frequency early in life in patients who have SWS is a prognostic indicator.[4]
Epidemiology
It occurs in approximately 1 in 50,000 newborns.[4]
Eponym
It is named for William Allen Sturge and Frederick Parkes Weber.[7][8][9]
Society and culture
The Sturge-Weber Foundation's (The SWF) international mission is to improve the quality of life and care for people with Sturge–Weber syndrome and associated port wine birthmark conditions. It supports affected individuals and their families with education, advocacy, and research to promote effective management and awareness. The SWF was founded by Kirk and Karen Ball, who began searching for answers after their daughter was diagnosed with Sturge–Weber syndrome at birth. The SWF was incorporated in the US in 1987 as an International 501(c)(3) non-profit organization. In 1992, the mission was expanded to include individuals with capillary vascular birthmarks, Klippel Trenaunay (KT) and Port Wine Birthmarks.[citation needed]
The Hemispherectomy Foundation was formed in 2008 to assist families with children who have Sturge–Weber syndrome and other conditions that require hemispherectomy.[10] The Brain Recovery Project was formed in 2011 to fund research and establish rehabilitation protocols to help children who have had hemispherectomy surgery reach their full potential.[citation needed]
Sturge Weber UK (SWUK), formerly Sturge-Weber Foundation UK, is a volunteer-run registered charity formed in 1990. The charity exists to support those affected by Sturge Weber syndrome, promote research into the condition and raise awareness of the condition amongst both public and professionals. The charity was instrumental in setting up a specialist Sturge Weber clinic at Great Ormond Street Hospital.[11]
References
- ↑ "The Sturge-Weber Foundation : Home". Archived from the original on 2011-11-29. Retrieved 2022-03-14.
- ↑ Shirley, Matthew D.; Tang, Hao; Gallione, Carol J.; Baugher, Joseph D.; Frelin, Laurence P.; Cohen, Bernard; North, Paula E.; Marchuk, Douglas A.; Comi, Anne M.; Pevsner, Jonathan (8 May 2013). "Sturge–Weber Syndrome and Port-Wine Stains Caused by Somatic Mutation in". New England Journal of Medicine. 368 (21): 1971–9. doi:10.1056/NEJMoa1213507. PMC 3749068. PMID 23656586.
- ↑ Zaki, Syed Ahmed; Vijay Lad (July 2011). "Sturge-Weber syndrome with bilateral facial nevus and early cerebral calcification". Journal of Pediatric Neurosciences. 6 (2): 114–5. doi:10.4103/1817-1745.92825 (inactive 31 May 2021). PMC 3296402. PMID 22408657.
{{cite journal}}: CS1 maint: DOI inactive as of May 2021 (link) - ↑ 4.0 4.1 4.2 4.3 Thomas-Sohl, Kristin A; Vaslow, Dale F; Maria, Bernard L (May 2004). "Sturge-Weber syndrome: A review". Pediatric Neurology. 30 (5): 303–310. doi:10.1016/j.pediatrneurol.2003.12.015. PMID 15165630.
- ↑ "Sturge-Weber syndrome: Radiopaedia.org". Archived from the original on 2011-11-27. Retrieved 2021-07-05.
- ↑ "Norfolk girl recovers after half of brain 'switched off'". BBC News. 2011-05-20. Archived from the original on 2021-05-25. Retrieved 2021-07-05.
- ↑ synd/1764 at Who Named It?
- ↑ Sturge WA (1879). "A case of partial epilepsy, apparently due to a lesion of one of the vasomotor centres of the brain". Transactions of the Clinical Society of London. 12: 162.
- ↑ Weber FP (1922). "Right-sided hemi-hypertrophy resulting from right-sided congenital spastic hemiplegia, with a morbid condition of the left side of the brain, revealed by radiograms". Journal of Neurology and Psychopathology. London. 3 (10): 134–9. doi:10.1136/jnnp.s1-3.10.134. PMC 1068054. PMID 21611493.
- ↑ "The Community News". Archived from the original on March 29, 2009. Retrieved 2009-02-25.
- ↑ "Search Results | Great Ormond Street Hospital". Archived from the original on 2013-12-30. Retrieved 2021-07-05.
Further reading
- Greenwood M, Meechan JG (July 2003). "General medicine and surgery for dental practitioners Part 4: Neurological disorders". Br Dent J. 195 (1): 19–25. doi:10.1038/sj.bdj.4810275. PMID 12856021.
Fig. 2 A patient with Sturge Weber Syndrome{{cite journal}}: External link in|quote=
External links
| Classification | |
|---|---|
| External resources |
- Pages with script errors
- CS1 maint: DOI inactive as of May 2021
- All articles with unsourced statements
- Articles with unsourced statements from December 2020
- Articles with invalid date parameter in template
- Articles with hatnote templates targeting a nonexistent page
- CS1 errors: external links
- Congenital disorders of eye, ear, face and neck
- Congenital disorders of nervous system
- Genodermatoses
- Rare syndromes
- Syndromes affecting the skin
- Syndromes affecting the nervous system
- Syndromes with mental retardation