Smith–Lemli–Opitz syndrome
| Smith–Lemli–Opitz syndrome | |
|---|---|
| Other names: SLOS, or 7-dehydrocholesterol reductase deficiency | |
 | |
| 7-Dehydrocholesterol is a toxic steroidal metabolite that accumulates in the bodies of those with SLOS | |
| Usual onset | Present at birth |
| Frequency | 1 in 20,000 to 1 in 60,000 |
Smith–Lemli–Opitz syndrome is an inborn error of cholesterol synthesis.[1] It is an autosomal recessive, multiple malformation syndrome caused by a mutation in the enzyme 7-Dehydrocholesterol reductase encoded by the DHCR7 gene. It causes a broad spectrum of effects, ranging from mild intellectual disability and behavioural problems to lethal malformations.[2]
Signs and symptoms
SLOS can present itself differently in different cases, depending on the severity of the mutation and other factors. Originally, SLOS patients were classified into two categories (classic and severe) based on physical and mental characteristics, alongside other clinical features. Since the discovery of the specific biochemical defect responsible for SLOS, patients are given a severity score based on their levels of cerebral, ocular, oral, and genital defects. It is then used to classify patients as having mild, classical, or severe SLOS.[3]
Physical characteristics
The most common facial features of SLOS include microcephaly, bitemporal narrowing (reduced distance between temples), ptosis, a short and upturned nose, micrognathia, epicanthal folds, and capillary hemangioma of the nose.[3] Other physical characteristics include:[citation needed]
- low-set and posteriorly rotated ears
- high-arched, narrow, hard palate
- cleft lip/palate
- agenesis or hypoplasia of the corpus callosum
- cerebellar hypoplasia
- increased ventricular size
- decreased frontal lobe size
- polydactyly of hands or feet
- short, proximally placed thumb
- other finger malformations
- syndactyly of second and third toes
- ambiguous or female-like male genitalia
- congenital heart defects
- renal, pulmonary, liver and eye abnormalities
-
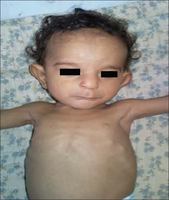
Large forehead, hypertelorism
-

Syndactyly of the second and third toes


Behavioural characteristics
Certain behaviours and attributes are commonly seen among patients suffering from SLOS. They may have low normal intelligence, and react negatively or with hypersensitivity to different sensory stimuli. This is particularly true for certain auditory and visual stimuli. Many patients show aggressiveness and self-injurious behaviours, and sleep disturbances are common.[3] Specific behaviours resembling those of people with autism are often present as well as hyperactivity, which provides genetic and biological insights into autism spectrum disorders. The autistic behaviours most characteristic of SLOS patients are opisthokinesis (an upper body movement), stretching of the upper body, and hand flicking.[4] Autism is typically diagnosed separately from SLOS using the DSM-V, and approximately 50–75% of SLOS patients meet the criteria for autism.[5]
Other behaviours associated with SLOS can be linked directly to physical abnormalities. For example, infants often show feeding problems or feeding intolerance, and patients may require increased caloric intake due to accelerated metabolism. Recurrent infections, including ear infections and pneumonia, are also common.[3]
Biochemical phenotype
Given that SLOS is caused by a mutation in an enzyme involved in cholesterol synthesis, the resulting biochemical characteristics may be predictable. Most patients have lowered plasma cholesterol levels (hypocholesterolemia). However, approximately 10% may show normal cholesterol levels,[3] and decreased concentrations of cholesterol are not solely indicative of SLOS. Increased levels of cholesterol precursors are also common in SLOS. In particular, elevated levels of 7-dehydrocholesterol are fairly specific to SLOS.[2]
Genetics
DHCR7
The gene encoding DHCR7 (labeled as DHCR7) was cloned in 1998, and has been mapped to chromosome 11q12-13.[1] It is 14100 base pairs of DNA in length, and contains nine exons,[2] the corresponding mRNA is 2786 base pairs in length (the remaining DNA sequence is intronic). The structure of the DHCR7 rat gene is very similar to the structure of the human gene.[1]
The highest levels of DHCR7 expression have been detected in the adrenal gland, the testis, the liver and in brain tissue.[1] Its expression is induced by decreased sterol concentrations via sterol regulatory binding proteins (SREBP). There is also evidence that its activity may be regulated by tissue specific transcription, and alternative splicing.[citation needed]
As outlined above, the enzyme DHCR7 catalyzes the reduction of 7DHC to cholesterol, as well as the reduction of 7-dehydrodesmosterol to desmosterol. It requires NADPH as a cofactor for this reduction, and may involve the activity of cytochrome-P450 oxidoreductase. It is also thought to contain iron.[1] DHCR7 is an integral membrane protein of the endoplasmic reticulum, and computer models have predicted up to nine transmembrane domains.[2] DHCR7 is most efficient at reducing 7DHC, but it is known to reduce the carbon 7 double bond of other sterols, indicating a range of substrate specificity. The human version of this enzyme is predicted to have a molecular weight of 54,489 kDa, and an isoelectric point of 9.05.[1]
The amino acid sequence that encodes DHCR7 is predicted to contain 475 amino acids, as well as several protein motifs. It contains multiple sterol reductase motifs, as would be expected given its function. It contains a potential sterol-sensing domain (SSD), whose function is unknown but thought to be necessary for binding sterol substrates. It also includes multiple sites of phosphorylation, including potential protein kinase C and tyrosine kinase sites (regulatory enzymes responsible for phosphorylation). The exact function of phosphorylating DHCR7 is yet unknown, but it is thought to be involved in the regulation of its activity.[1]
Mutations and incidence

SLOS is an autosomal recessive disorder.[6] More than 130 different types of mutations have been identified.[2] Missense mutations (single nucleotide change resulting in a code for a different amino acid) are the most common, accounting for 87.6% of the SLOS spectrum. These typically reduce the function of the enzyme but may not inhibit it completely. Much depends on the nature of the mutation (i.e. which amino acid is replaced and where). Null mutations are much less common, these mutations produce either a completely dysfunctional enzyme, or no enzyme at all.[6] Thus, missense mutations may be more common overall because they are less lethal than nonsense mutations; nonsense mutations may simply result in spontaneous abortion.[citation needed]
The IVS8-1G>C is the most frequently reported mutation in DHCR7. This disrupts the joining of exons eight and nine, and results in the insertion of 134 nucleotides into the DHCR7 transcript. This is a nonsense mutation, thus patients that are homozygous for this allele are severely affected. It is thought that this mutation first occurred in the British Isles, and it has a carrier (those that are heterozygous for the allele but not affected) frequency of 1.09% for Caucasians of European heritage. The frequency of mutations differs for various ethnicities, depending on the origin of the mutation. In all Caucasian populations, this particular mutation has an estimated carrier frequency of 3%.[1]
The next most common mutation is 278C>T, and results in a threonine at the amino acid position 93. It is a missense mutation and tends to be associated with less severe symptoms. This mutation is the most common one seen in patients of Italian, Cuban, and Mediterranean descent.[1]
The third most common mutation is 452G>A. This nonsense mutation causes protein termination, such that the enzyme DHCR7 would not be formed. It is thought to have arisen in Southern Poland and is most common in Northern Europe.[1]
Other mutations are less common, although appear to target certain protein domains more so than others. For example, the sterol reductase motifs are common sites of mutation.[1] Overall, there is an estimated carrier frequency (for any DHCR7 mutation causing SLOS) of 3-4% in Caucasian populations (it is less frequent among Asian and African populations[7]). This number indicates a hypothetical birth incidence between 1/2500 and 1/4500. However, the measured incidence is between 1/10,000 to 1/60,000 (it differs depending on heritage and descent).[6] This is much lower than expected. This indicates that many cases of SLOS are undetected, and is likely due to either spontaneous abortion caused by severe mutations (miscarriage), or mild cases that are undiagnosed. Females lack the characteristic genital malformations that affected males have, and thus are less likely to be correctly diagnosed.[7]
Cholesterol metabolism and function
Metabolism
Cholesterol can be obtained through the diet, but it can also be formed by metabolism in the body. Cholesterol metabolism primarily takes place in the liver, with significant amounts in the intestine as well.[8] It should also be noted that cholesterol cannot pass the blood–brain barrier, thus within the brain, biosynthesis is the only source of cholesterol.[9]

In humans, cholesterol synthesis begins with the mevalonate pathway (see diagram), leading to the synthesis of farnesyl pyrophosphate (FPP). This pathway uses two acetyl-CoA and two NADPH to make mevalonate, which is metabolized to isopentenyl pyrophosphate (IPP) using three ATP. From there, three IPP are needed to make one FPP. The combination of two FPP leads to the formation of squalene; this represents the first committed step towards cholesterol biosynthesis.[10] Squalene leads to the creation of lanosterol, from which there are multiple pathways that lead to cholesterol biosynthesis. The rate limiting step of cholesterol synthesis is the conversion of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) to mevalonate, this is an early step in the mevalonate pathway catalyzed by HMG-CoA reductase.[citation needed]


Through a complicated series of reactions, lanosterol leads to the formation of zymosterol. As shown in a diagram to the right, it is at this point that the pathway diverges. In humans, the main pathway leading to cholesterol is known as the Kandutsch–Russell pathway.[3] Zymosterol is metabolized to 5α-cholesta-7,24-dien-3β-ol, then to lathosterol, and then to 7-dehydrocholesterol, or 7-DHC. 7-DHC is the immediate precursor to cholesterol, and the enzyme DHCR7 is responsible for converting 7-DHC to cholesterol.[1] DHCR7 reduces the double bond on carbon 7 of 7-DHC, leading to the unesterified product.[9] Mutations in this enzyme are responsible for the wide range of defects present in SLOS. In another pathway leading to cholesterol synthesis, DHCR7 is required for the reduction of 7-Dehydrodesmosterol to desmosterol.[citation needed]
Regulation
Regulation of cholesterol synthesis is complex and occurs primarily through the enzyme HMG-CoA reductase (catalyst of the rate-limiting step). It involves a feedback loop that is sensitive to cellular levels of cholesterol. The four main steps of regulation are:[8]
- The synthesis of the enzyme HMG-CoA reductase is controlled by sterol regulatory element binding protein (SREBP). This is a transcription factor that is inactive when cholesterol levels are high, and active when cholesterol levels are low. When cholesterol levels fall, SREBP is released from the nuclear membrane or endoplasmic reticulum, it then migrates to the nucleus and causes the transcription of the HMG-CoA reductase gene.
- The translation (creating the enzyme from the mRNA transcript) of HMG-CoA reductase is inhibited by derivatives of mevalonate and by dietary cholesterol.
- The degradation of HMG-CoA reductase is tightly controlled. The part of the enzyme that is bound to the endoplasmic reticulum senses signals, such as increased cholesterol levels, that lead to its degradation or proteolysis.
- When HMG-CoA reductase is phosphorylated, its activity decreases. This means cholesterol synthesis is reduced when cell energy (ATP) levels are low.
Function
Cholesterol is an important lipid involved in metabolism, cell function, and structure. It is a structural component of the cell membrane,[1] such that it provides structure and regulates the fluidity of the phospholipid bilayer. Furthermore, cholesterol is a constituent in lipid rafts. These are congregations of proteins and lipids (including sphingolipids and cholesterol) that float within the cell membrane, and play a role in the regulation of membrane function. Lipid rafts are more ordered or rigid than the membrane bilayer surrounding them. Their involvement in regulation stems mostly from their association with proteins; upon binding substrates, some proteins have a higher affinity for attaching to lipid rafts. This brings them in close proximity with other proteins, allowing them to affect signaling pathways. Cholesterol specifically acts as a spacer and a glue for lipid rafts; absence of cholesterol leads to the dissociation of proteins.[11]
Given its prevalence in cell membranes, cholesterol is highly involved in certain transport processes. It may influence the function of ion channels and other membrane transporters. For example, cholesterol is necessary for the ligand binding activity of the serotonin receptor.[12] In addition, it appears to be very important in exocytosis. Cholesterol modulates the properties of the membrane (such as membrane curvature), and may regulate the fusion of vesicles with the cell membrane. It may also facilitate the recruitment of complexes necessary for exocytosis. Given that neurons rely heavily on exocytosis for the transmission of impulses, cholesterol is a very important part of the nervous system.[13]

One particularly relevant pathway in which cholesterol takes place is the Hedgehog signaling pathway. This pathway is very important during embryonic development, and involved in deciding the fate of cells (i.e., which tissue they need to migrate to). Hedgehog proteins are also involved in the transcription of genes that regulate cell proliferation and differentiation. Cholesterol is important to this pathway because it undergoes covalent bonding to Hedgehog proteins, resulting in their activation. Without cholesterol, the signaling activity is disrupted and cell differentiation may be impaired.[14]
Cholesterol is a precursor for many important molecules. These include bile acids (important in processing dietary fats), oxysterols, neurosteroids (involved in neurotransmission and excitation), glucocorticoids (involved in immune and inflammatory processes), mineralocorticoids (osmotic balance), and sex steroids (i.e. estrogen and testosterone; wide range of function but involved in genital development prenatally).[1] Finally, cholesterol is a major component of myelin, a protective layer around neurons. Myelination occurs most rapidly during prenatal development, meaning that the demand for cholesterol biosynthesis is very high.[9]
Pathogenesis
Given that the function of cholesterol encompasses a very wide range, it is unlikely that the symptoms of SLOS are due to a single molecular mechanism. Some of the molecular effects are yet unknown, but could be extrapolated based on the role of cholesterol. In general, the negative effects are due to decreased levels of cholesterol and increased levels of cholesterol precursors-most notably, 7DHC. Although 7DHC is structurally similar to cholesterol, and could potentially act as a substitute, the effects of this are still being studied.[2]
Most patients with SLOS present decreased cholesterol levels, particularly in the brain (where cholesterol levels rely primarily on new synthesis). This also means that any sterol derivatives of cholesterol would also have reduced concentrations. For example, reduced levels of neurosteroids may be seen in SLOS. These are lipids which take part in signaling within the brain, and must be produced within the brain itself. They are responsible for interacting with nuclear steroid receptors, and bind to neurotransmitter-gated ion channels. Specifically, they modulate the effects of GABA and NMDA receptors, resulting in calming effects, improved memory, and more. Thus, given that some characteristics of SLOS are the opposite of these effects (hyperactivity, anxiety), a reduction in neurosteroids could influence both neurological development and behaviour.[15]

Furthermore, as outlined above, cholesterol is an important aspect in Hedgehog signaling. With lower levels of cholesterol, hedgehog proteins would not undergo the necessary covalent modification and subsequent activation. This would result in impaired embryonic development, and may contribute to the observed physical birth defects in SLOS. One particular hedgehog signaling protein, sonic hedgehog (SHH), is important in the pattern of the central nervous system, facial features, and limbs.[2] Other hedgehog proteins may be involved in the development of the genital tract and the skeleton.[3]
The altered sterol levels in SLOS are particularly relevant to cell membranes, which are made primarily of lipids. SLOS patients may show cell membranes with abnormal properties or composition, and reduced cholesterol levels greatly affect the stability and proteins of lipid rafts.[2] Despite their structural similarity, 7DHC is unable so replace cholesterol in lipid rafts.[16] In addition, a lack of cholesterol contributes to the increased fluidity of the cell membrane, and may cause abnormal granule secretions.[2] All of these changes in the membrane likely contribute to changes in transport functions that are observed in SLOS. They may cause defects in IgE receptor-mediated mast cell degranulation and cytokine production, which are cells involved in allergic and immune responses.[2] The NMDA receptor is affected, as well as the binding capability of the hippocampal serotonin receptor.[12] Cell to cell interaction, which is very important in development, may be impaired.[3] Exocytosis in synaptic vesicles has been shown to be reduced, likely due to impaired vesicle fusion to the cell membrane, or poor vesicle recycling.[13] Finally, cholesterol is highly prevalent in myelin, therefore SLOS patients show reduced myelination of the cerebral hemispheres, peripheral nerves, and cranial nerves.[15]
In addition to lowered levels of cholesterol, many of the symptoms shown in SLOS stem from the toxic effects of 7DHC. 7DHC is known to impair intracellular cholesterol transport. It also increases the degradation of HMG-CoA reductase (the enzyme that catalyzes the rate-limiting step in cholesterol synthesis). 7DHC leads to novel oxysterol and steroid derivatives, and many of their functions or effects are yet unknown.[2] A very important finding with respect to 7DHC is that it is the most reactive lipid for lipid peroxidation, and results in systemic oxidative stress. Lipid peroxidation is known to destroy membranes of both cells and membrane-bound organelles. The derivative of 7DHC that is used to indicate oxidative stress is 3β,5α-dihydroxy-cholest-7-en-6-one (DHCEO); it is formed from a primary product of 7DHC peroxidation, 7-DHC-5α,6α-epoxide. DHCEO is toxic to cortical neuronal and glial cells, and accelerates their differentiation and arborization.[17] Through oxidative stress, 7DHC is thought to be responsible for the increased photosensitivity shown in SLOS patients. Normal UVA exposure may lead to oxidative stress in skin cells. Given that 7DHC is more readily oxidized, it enhances the effects of UVA, leading to increased membrane lipid oxidation and increased production of reactive oxygen species (ROS).[16]
Typically, more altered the levels of 7DHC and cholesterol lead to more severe symptoms of SLOS. The levels of these metabolites also correspond to the severity of the mutation (nonsense versus missense); some mutations of DHCR7 may still show residual cholesterol synthesis, and others may not. However, even individuals with the same mutations or genotype may still show variability in their symptoms. This may be due to maternal factors, such as the transfer of cholesterol to the fetus during pregnancy, as well as the amount of cholesterol present in the brain before the blood–brain barrier forms prenatally. The rate of accumulation and excretion of toxic metabolites may vary from person to person. Maternal apolipoprotein E has also been implicated in individual variability in SLOS, although the exact nature of this relationship is unknown.[6] There are likely more factors contributing to the wide spectrum of effects in SLOS which have not yet been discovered.[citation needed]
Screening and diagnosis
Prenatally
The most characteristic biochemical indicator of SLOS is an increased concentration of 7DHC (reduced cholesterol levels are also typical, but appear in other disorders as well). Thus, prenatally, SLOS is diagnosed upon finding an elevated 7DHC:total sterol ratio in fetal tissues, or increased levels of 7DHC in amniotic fluid. The 7DHC:total sterol ratio can be measured at 11–12 weeks of gestation by chorionic villus sampling, and elevated 7DHC in amniotic fluid can be measured by 13 weeks. Furthermore, if parental mutations are known, DNA testing of amniotic fluid or chorionic villus samples may be performed.[3]

Amniocentesis (process of sampling amniotic fluid) and chorionic villus sampling cannot be performed until approximately 3 months into the pregnancy. Given that SLOS is a very severe syndrome, parents may want to choose to terminate their pregnancy if their fetus is affected. Amniocentesis and chorionic villus sampling leave very little time to make this decision (abortions become more difficult as the pregnancy advances), and can also pose severe risks to the mother and baby. Thus, there is a very large desire for noninvasive midgestation diagnostic tests.[18] Examining the concentrations of sterols in maternal urine is one potential way to identify SLOS prenatally. During pregnancy, the fetus is solely responsible for synthesizing the cholesterol needed to produce estriol. A fetus with SLOS cannot produce cholesterol, and may use 7DHC or 8DHC as precursors for estriol instead. This creates 7- or 8-dehydrosteroids (such as 7-dehydroestriol), which may show up in the maternal urine. These are novel metabolites due to the presence of a normally reduced double bond at carbon 7 (caused by the inactivity of DHCR7), and may be used as indicators of SLOS.[19] Other cholesterol derivatives which possess a double bond at the 7th or 8th position and are present in maternal urine may also be indicators of SLOS. 7- and 8-dehydropregnanetriols have been shown to be present in the urine of mothers with an affected fetus but not with an unaffected fetus, and thus are used in diagnosis. These pregnadienes originated in the fetus and traveled through the placenta before reaching the mother. Their excretion indicates that neither the placenta nor the maternal organs have necessary enzymes needed to reduce the double bond of these novel metabolites.[18]
Postnatally
If SLOS goes undetected until after birth, diagnosis may be based on the characteristic physical features as well as finding increased plasma levels of 7DHC.[citation needed]
There are many different ways of detecting 7DHC levels in blood plasma, one way is using the Liebermann–Burchard (LB) reagent. This is a simple colorimetric assay developed with the intention of use for large scale screening. When treated with the LB reagent, SLOS samples turn pink immediately and gradually become blue; normal blood samples are initially colorless and develop a faint blue color. Although this method has limitations and is not used to give a definitive diagnosis, it has appeal in that it is a much faster method than using cell cultures.[20]
Another way of detecting 7DHC is through gas chromatography, a technique used to separate and analyze compounds. Selected ion monitoring gas chromatography/mass-spectrometry (SIM-GC/MS) is a very sensitive version of gas chromatography, and permits detection of even mild cases of SLOS.[21] Other methods include time-of-flight mass spectrometry, particle-beam LC/MS, electrospray tandem MS, and ultraviolet absorbance, all of which may be used on either blood samples, amniotic fluid, or chorionic villus. Measuring levels of bile acids in patients urine, or studying DCHR7 activity in tissue culture are also common postnatal diagnostic techniques.[20]
Treatment
Management of individuals with SLOS is complex and often requires a team of specialists. Some of the congenital malformations (cleft palate) can be corrected with surgery.[7] Other treatments have yet to be proven successful in randomized studies, however anecdotally they appear to cause improvements.[citation needed]
Cholesterol supplementation
Currently, the most common form of treatment for SLOS involves dietary cholesterol supplementation.[22] Anecdotal reports indicate that this has some benefits; it may result in increased growth, lower irritability, improved sociability, less self-injurious behaviour, less tactile defensiveness, fewer infections, more muscle tone, less photosensitivity and fewer autistic behaviours.[23] Cholesterol supplementation begins at a dose of 40–50 mg/kg/day, increasing as needed. It is administered either through consuming foods high in cholesterol (eggs, cream, liver), or as purified food grade cholesterol. Younger children and infants may require tube feeding.[3] However, dietary cholesterol does not reduce the levels of 7DHC, cannot cross the blood–brain barrier, and does not appear to improve developmental outcomes.[23] One empirical study found that cholesterol supplementation did not improve developmental delay, regardless of the age at which it began. This is likely because most developmental delays stem from malformations of the brain, which dietary cholesterol cannot ameliorate due to its inability to cross the blood–brain barrier.[24]

Simvastatin therapy
HMG-CoA reductase inhibitors have been examined as treatment for SLOS. Given that this catalyzes the rate-limiting step in cholesterol synthesis, inhibiting it would reduce the buildup of toxic metabolites such as 7DHC.[22] Simvastatin is a known inhibitor of HMG-CoA reductase, and most importantly is able to cross the blood–brain barrier. It has been reported to decrease the levels of 7DHC, as well as increase the levels of cholesterol.[23] The increased cholesterol levels are due to simvastatin's effect on the expression of different genes. Simvastatin increases the expression of DHCR7, likely leading to increased activity of DHCR7. It has also been shown to increase the expression of other genes involved in cholesterol synthesis and uptake. However, these benefits are dependent on the amount of residual cholesterol synthesis. Because some individuals possess less severe mutations and demonstrate some amount of DCHR7 activity, these people benefit the most from simvastatin therapy as they still have a partially functioning enzyme. For individuals that show no residual DCHR7 activity, such as those homozygous for null alleles or mutations, simvastatin therapy may actually be toxic.[22] This highlights the importance of identifying the specific genotype of the SLOS patient before administering treatment. It is still unknown if simvastatin will improve the behavioural or learning deficits in SLOS.[23]
Antioxidant supplementation
Antioxidants are those which inhibit the oxidation of molecules or reduce metabolites that were previously oxidized. Given that some symptoms of SLOS are thought to result from the peroxidation of 7DHC and its derivatives, inhibiting this peroxidation would likely have beneficial effects. Antioxidants have been shown to increase the level of lipid transcripts in SLOS cells, these transcripts play a role in lipid (cholesterol) biosynthesis and are known to be down-regulated in SLOS. Furthermore, vitamin E specifically is known to decrease DHCEO levels, which is an indicator of oxidative stress in SLOS, as well as present beneficial changes in gene expression. Vitamin E appears to be the most powerful antioxidant for treating SLOS, and in mouse models has reduced the levels of oxysterols in the brain. However, antioxidants have only been studied in animal models of SLOS or isolated SLOS cells. Thus, their clinical significance and negative side effects are still unknown, and their use has yet to be studied in humans.[25]
Further considerations
When treating SLOS, a recurring issue is whether or not the intellectual and behavioural deficits are due to fixed developmental problems (i.e. fixed brain malformations), or due to ongoing abnormal sterol levels that interrupt the normal function of the brain and other tissues.[22] If the latter is true, then treatments which change the sterol levels and ratios, particularly in the brain, will likely improve the developmental outcome of the patient. However, if the former is true, then treatment is likely to help only with symptoms and not with specific developmental deficits.[citation needed]
Research
The most common animal used to study SLOS is the mouse. According to BioCyc, cholesterol biosynthesis in mice is very similar to that of humans. Most importantly, mice possess both DHCR7 (the enzyme responsible for SLOS), and HMG-CoA reductase (the rate limiting step of cholesterol synthesis.[26] Rats are similar to mice and have also been used. There are two popular ways in which animal models of SLOS are created. The first is using teratogens, the second is using genetic manipulations to create mutations in the DHCR7 gene.[citation needed]
Teratogenic models
Teratogenic models are induced by feeding pregnant rats or mice inhibitors of DCHR7. Two common inhibitors are BM15766 (4-(2-[1-(4-chlorocinnamyl)piperazin-4-yl]ethyl)-benzoic acid) and AY9944 (trans-l,4-bis(2-chlorobenzylaminomethy1)cyclohexane dihydrochloride). These compounds have different chemical and physical properties, but induce similar effects. AY9944 has been shown to induce holoprosencephaly and sexual malformations similar to those seen in humans with SLOS.[27] It is also known to cause impairments in the serotonin receptor, another defect commonly seen in SLOS patients.[28] BM15766 has produced the lack of cholesterol and bile acid synthesis that is seen in SLOS patients with homozygous mutations. All teratogenic models can be effectively used to study SLOS; however, they present lower levels of 7-DHC and 8-DHC than are seen in humans. This can be explained by the fact that humans experience a permanent block in their DHCR7 activity, where mice and rats treated with inhibitors experience only transient blocks. Furthermore, different species of mice and rats are more resistant to teratogens, and may be less effective as models of SLOS.[27] Teratogenic models are most commonly used to study more long-term effects of SLOS, because they survive longer than genetic models. For example, one study examined the retinal degeneration of SLOS, which in rats does not occur until at least one month after birth.[28]
Genetic models
Genetic models of SLOS are created by knocking out the DHCR7 gene. One study used homologous recombination to disrupt DCHR7 in mouse embryonic stem cells. Similar to what is found in humans, heterozygous mice (having only one mutated allele) were phentoypically normal, and were crossed to produce pups (young mice) homozygous for the mutated allele. Although these pups died within the first day of life due to their inability to feed, they showed characteristics similar to humans with SLOS. They had decreased levels of cholesterol, increased levels of 7- and 8DHC, showed less growth and smaller birth weights, had craniofacial malformations, and less movement. Many also had a cleft palate, and decreased neuronal responses to glutamate. Overall however, the pups had fewer dysmorphic features than human patients with SLOS; they did not present limb, renal, adrenal or central nervous system malformations. This is explained by the fact that in rodents, maternal cholesterol can cross the placenta, and actually appears to be essential for the development of the fetus. In humans, very little maternal cholesterol is transferred to the fetus. In sum, the genetic mouse model is helpful to explain the neuropathophysiology of SLOS.[29]
Discoveries
Many discoveries in SLOS research have been made using animal models. They have been used to study different treatment techniques, including the effectiveness of simvastatin therapy.[23] Other studies have examined behavioural characteristics while attempting to explain their underlying pathogenesis.[30] A common finding is that mouse models of SLOS show abnormal serotonergic development, which may be at least partially responsible for the autistic behaviours seen in SLOS.[31] Mouse models have also been used to develop diagnostic techniques; multiple studies have examined biomarkers that result from the oxidation of 7DHC, such as DHCEO.[17][32] It is likely that as animal models are improved, they will lead to many more discoveries in SLOS research.[citation needed]
Eponym
It is named after David Weyhe Smith (1926–1981), an American pediatrician; Luc Lemli (1935–), a Belgian physician; and John Marius Opitz (1935–), a German-American physician. These are the researchers who first described the symptoms of SLOS.[33]
References
- ↑ 1.00 1.01 1.02 1.03 1.04 1.05 1.06 1.07 1.08 1.09 1.10 1.11 1.12 1.13 Correa-Cerro, Lina S.; Porter, Forbes D. (2005). "3β-Hydroxysterol Δ7-reductase and the Smith–Lemli–Opitz syndrome". Molecular Genetics and Metabolism. 84 (2): 112–26. doi:10.1016/j.ymgme.2004.09.017. PMID 15670717. Archived (PDF) from the original on 2021-08-29. Retrieved 2021-08-11.
- ↑ 2.00 2.01 2.02 2.03 2.04 2.05 2.06 2.07 2.08 2.09 2.10 Porter, Forbes D (2008). "Smith–Lemli–Opitz syndrome: Pathogenesis, diagnosis and management". European Journal of Human Genetics. 16 (5): 535–41. doi:10.1038/ejhg.2008.10. PMID 18285838. Archived from the original on 2019-09-28. Retrieved 2021-08-11.
- ↑ 3.0 3.1 3.2 3.3 3.4 3.5 3.6 3.7 3.8 3.9 Nowaczyk, MJM; Waye, JS (2001). "The Smith-Lemli-Opitz syndrome: A novel metabolic way of understanding developmental biology, embryogenesis, and dysmorphology". Clinical Genetics. 59 (6): 375–86. doi:10.1034/j.1399-0004.2001.590601.x. PMID 11453964. S2CID 9146017.
- ↑ Ghaziuddin, Mohammad; Al-Owain, Mohammed (2013). "Autism Spectrum Disorders and Inborn Errors of Metabolism: An Update". Pediatric Neurology. 49 (4): 232–6. doi:10.1016/j.pediatrneurol.2013.05.013. PMID 23921282.
- ↑ Bukelis, I.; Porter, F. D.; Zimmerman, A. W.; Tierney, E. (2007). "Smith-Lemli-Opitz Syndrome and Autism Spectrum Disorder". American Journal of Psychiatry. 164 (11): 1655–61. doi:10.1176/appi.ajp.2007.07020315. PMID 17974928.
- ↑ 6.0 6.1 6.2 6.3 Yu, H; Patel, SB (2005). "Recent insights into the Smith-Lemli-Opitz syndrome". Clinical Genetics. 68 (5): 383–91. doi:10.1111/j.1399-0004.2005.00515.x. PMC 1350989. PMID 16207203.
- ↑ 7.0 7.1 7.2 Nowaczyk, Malgorzata JM (June 20, 2013). "Smith-Lemli-Opitz Syndrome". In Pagon, Roberta A; Adam, Margaret P; Bird, Thomas D; Dolan, Cynthia R; Fong, Chin-To; Smith, Richard JH; Stephens, Karen (eds.). GeneReviews. National Library of Medicine. Archived from the original on January 16, 2014. Retrieved December 5, 2013.
- ↑ 8.0 8.1 Berg, Jeremy M; Tymoczko, John L; Stryer, Lubert (2002). "The Complex Regulation of Cholesterol Biosynthesis Takes Place at Several Levels". Biochemistry (5th ed.). New York: W H Freeman. Section 26.3. ISBN 978-0-7167-3051-4.
- ↑ 9.0 9.1 9.2 Patti, G.J.; Shriver, L.P.; Wassif, C.A.; Woo, H.K.; Uritboonthai, W.; Apon, J.; Manchester, M.; Porter, F.D.; Siuzdak, G. (2010). "Nanostructure-initiator mass spectrometry (NIMS) imaging of brain cholesterol metabolites in Smith-Lemli-Opitz syndrome". Neuroscience. 170 (3): 858–64. doi:10.1016/j.neuroscience.2010.07.038. PMC 2952448. PMID 20670678.
- ↑ Liscum, Laura (2008). "Cholesterol biosynthesis". In Vance, D. E.; Vance, J. E. (eds.). Biochemistry of Lipids, Lipoproteins and Membranes (5th ed.). Elsevier. pp. 399–422. ISBN 978-0-08-055988-9.
- ↑ Simons, Kai; Ehehalt, Robert (2002). "Cholesterol, lipid rafts, and disease". Journal of Clinical Investigation. 110 (5): 597–603. doi:10.1172/JCI16390. PMC 151114. PMID 12208858.
- ↑ 12.0 12.1 Singh, Pushpendra; Paila, Yamuna Devi; Chattopadhyay, Amitabha (2007). "Differential effects of cholesterol and 7-dehydrocholesterol on the ligand binding activity of the hippocampal serotonin1A receptor: Implications in SLOS". Biochemical and Biophysical Research Communications. 358 (2): 495–9. doi:10.1016/j.bbrc.2007.04.135. PMID 17493586.
- ↑ 13.0 13.1 Linetti, A.; Fratangeli, A.; Taverna, E.; Valnegri, P.; Francolini, M.; Cappello, V.; Matteoli, M.; Passafaro, M.; Rosa, P. (2010). "Cholesterol reduction impairs exocytosis of synaptic vesicles". Journal of Cell Science. 123 (4): 595–605. doi:10.1242/jcs.060681. PMID 20103534.
- ↑ Ingham, Philip W. (2008). "Hedgehog signalling". Current Biology. 18 (6): R238–41. doi:10.1016/j.cub.2008.01.050. PMID 18364223.
- ↑ 15.0 15.1 Marcos, Josep; Guo, Li-Wei; Wilson, William K; Porter, Forbes D; Shackleton, Cedric (2004). "The implications of 7-dehydrosterol-7-reductase deficiency (Smith–Lemli–Opitz syndrome) to neurosteroid production". Steroids. 69 (1): 51–60. doi:10.1016/j.steroids.2003.09.013. PMID 14715377. S2CID 900728.
- ↑ 16.0 16.1 Valencia, Antonio; Rajadurai, Anpuchchelvi; Carle, A. Bjorn; Kochevar, Irene E. (2006). "7-Dehydrocholesterol enhances ultraviolet A-induced oxidative stress in keratinocytes: Roles of NADPH oxidase, mitochondria, and lipid rafts". Free Radical Biology and Medicine. 41 (11): 1704–18. doi:10.1016/j.freeradbiomed.2006.09.006. PMC 1880892. PMID 17145559.
- ↑ 17.0 17.1 Korade, Zeljka; Xu, Libin; Mirnics, Karoly; Porter, Ned A. (2012). "Lipid biomarkers of oxidative stress in a genetic mouse model of Smith-Lemli-Opitz syndrome". Journal of Inherited Metabolic Disease. 36 (1): 113–22. doi:10.1007/s10545-012-9504-z. PMC 3674764. PMID 22718275.
- ↑ 18.0 18.1 Shackleton, C; Roitman, E; Kratz, LE; Kelley, RI (1999). "Midgestational maternal urine steroid markers of fetal Smith–Lemli–Opitz (SLO) syndrome (7-dehydrocholesterol 7-reductase deficiency)". Steroids. 64 (7): 446–52. doi:10.1016/S0039-128X(99)00026-4. PMID 10443900. S2CID 42834575.
- ↑ Matabosch, Xavier; Rahman, Mahbuba; Hughes, Beverly; Patel, Shailendra B.; Watson, Gordon; Shackleton, Cedric (2009). "Steroid production and excretion by the pregnant mouse, particularly in relation to pregnancies with fetuses deficient in Δ7-sterol reductase (Dhcr7), the enzyme associated with Smith–Lemli–Opitz syndrome". The Journal of Steroid Biochemistry and Molecular Biology. 116 (1–2): 61–70. doi:10.1016/j.jsbmb.2009.04.011. PMC 2929956. PMID 19406241.
- ↑ 20.0 20.1 Xiong, Quanbo; Ruan, Benfang; Whitby, Frank G.; Tuohy, Richard P.; Belanger, Thomas L.; Kelley, Richard I.; Wilson, William K.; Schroepfer, George J. (2002). "A colorimetric assay for 7-dehydrocholesterol with potential application to screening for Smith–Lemli–Opitz syndrome". Chemistry and Physics of Lipids. 115 (1–2): 1–15. doi:10.1016/S0009-3084(01)00205-5. PMID 12047895.
- ↑ Kelley, Richard I. (1995). "Diagnosis of Smith-Lemli-Opitz syndrome by gas chromatography/mass spectrometry of 7-dehydrocholesterol in plasma, amniotic fluid and cultured skin fibroblasts". Clinica Chimica Acta. 236 (1): 45–58. doi:10.1016/0009-8981(95)06038-4. PMID 7664465.
- ↑ 22.0 22.1 22.2 22.3 Wassif, Christopher A.; Krakowiak, Patrycja A.; Wright, Brooke S.; Gewandter, Jennifer S.; Sterner, Allison L.; Javitt, Norman; Yergey, Alfred L.; Porter, Forbes D. (2005). "Residual cholesterol synthesis and simvastatin induction of cholesterol synthesis in Smith–Lemli–Opitz syndrome fibroblasts". Molecular Genetics and Metabolism. 85 (2): 96–107. doi:10.1016/j.ymgme.2004.12.009. PMID 15896653. Archived (PDF) from the original on 2021-08-29. Retrieved 2021-08-11.
- ↑ 23.0 23.1 23.2 23.3 23.4 Correa-Cerro, L. S.; Wassif, CA; Kratz, L; Miller, GF; Munasinghe, JP; Grinberg, A; Fliesler, SJ; Porter, FD (2006). "Development and characterization of a hypomorphic Smith-Lemli-Opitz syndrome mouse model and efficacy of simvastatin therapy". Human Molecular Genetics. 15 (6): 839–51. doi:10.1093/hmg/ddl003. PMID 16446309.
- ↑ Sikora, Darryn M; Ruggiero, Mark; Petit-Kekel, Kersti; Merkens, Louise S; Connor, William E; Steiner, Robert D (2004). "Cholesterol supplementation does not improve developmental progress in Smith-Lemli-Opitz syndrome". The Journal of Pediatrics. 144 (6): 783–91. doi:10.1016/j.jpeds.2004.02.036. PMID 15192627.
- ↑ Korade, Zeljka; Xu, Libin; Harrison, Fiona E.; Ahsen, Refayat; Hart, Sarah E.; Folkes, Oakleigh M.; Mirnics, Károly; Porter, Ned A. (2013). "Antioxidant Supplementation Ameliorates Molecular Deficits in Smith-Lemli-Opitz Syndrome". Biological Psychiatry. 75 (3): 215–22. doi:10.1016/j.biopsych.2013.06.013. PMC 3874268. PMID 23896203.
- ↑ Karp, P. D.; Ouzounis, CA; Moore-Kochlacs, C; Goldovsky, L; Kaipa, P; Ahrén, D; Tsoka, S; Darzentas, N; Kunin, V; López-Bigas, N (2005). "Expansion of the BioCyc collection of pathway/genome databases to 160 genomes". Nucleic Acids Research. 33 (19): 6083–9. doi:10.1093/nar/gki892. PMC 1266070. PMID 16246909.
- ↑ 27.0 27.1 Wolf, Claude; Chevy, Francoise; Pham, Jacques; Kolf-Clauw, Martine; Citadelle, Daniele; Mulliez, Nicole; Roux, Charles (1996). "Changes in serum sterols of rats treated with 7-dehydrocholesterol-Δ7-reductase inhibitors: Comparison to levels in humans with Smith-Lemli-Opitz syndrome". Journal of Lipid Research. 37 (6): 1325–33. doi:10.1016/S0022-2275(20)39162-8. PMID 8808767.
{{cite journal}}: CS1 maint: url-status (link) - ↑ 28.0 28.1 Xu, Libin; Sheflin, Lowell G.; Porter, Ned A.; Fliesler, Steven J. (2012). "7-Dehydrocholesterol-derived oxysterols and retinal degeneration in a rat model of Smith–Lemli–Opitz syndrome". Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 1821 (6): 877–83. doi:10.1016/j.bbalip.2012.03.001. PMC 3340457. PMID 22425966.
- ↑ Wassif, C. A.; Zhu, P; Kratz, L; Krakowiak, PA; Battaile, KP; Weight, FF; Grinberg, A; Steiner, RD; Nwokoro, NA; Kelley, RI; Stewart, RR; Porter, FD (2001). "Biochemical, phenotypic and neurophysiological characterization of a genetic mouse model of RSH/Smith-Lemli-Opitz syndrome". Human Molecular Genetics. 10 (6): 555–64. doi:10.1093/hmg/10.6.555. PMID 11230174.
- ↑ Korade, Z.; Folkes, O.M.; Harrison, F.E. (2013). "Behavioral and serotonergic response changes in the Dhcr7-HET mouse model of Smith–Lemli–Opitz syndrome". Pharmacology Biochemistry and Behavior. 106: 101–8. doi:10.1016/j.pbb.2013.03.007. PMID 23541496. S2CID 38380298.
- ↑ Waage-Baudet, H; Lauder, J.M; Dehart, D.B; Kluckman, K; Hiller, S; Tint, G.S; Sulik, K.K (2003). "Abnormal serotonergic development in a mouse model for the Smith–Lemli–Opitz syndrome: Implications for autism". International Journal of Developmental Neuroscience. 21 (8): 451–9. doi:10.1016/j.ijdevneu.2003.09.002. PMID 14659996. S2CID 42098029.
- ↑ Xu, L.; Korade, Z.; Rosado, D. A.; Liu, W.; Lamberson, C. R.; Porter, N. A. (2011). "An oxysterol biomarker for 7-dehydrocholesterol oxidation in cell/mouse models for Smith-Lemli-Opitz syndrome". The Journal of Lipid Research. 52 (6): 1222–33. doi:10.1194/jlr.M014498. PMC 3090243. PMID 21402677.
- ↑ Smith, David W.; Lemli, Luc; Opitz, John M. (1964). "A newly recognized syndrome of multiple congenital anomalies". The Journal of Pediatrics. 64 (2): 210–7. doi:10.1016/S0022-3476(64)80264-X. PMID 14119520.
![]() This article incorporates public domain material from the United States National Library of Medicine document: "Genetics Home Reference".
This article incorporates public domain material from the United States National Library of Medicine document: "Genetics Home Reference".
External links
| Classification | |
|---|---|
| External resources |
- GeneReview/UW/NIH on Smith–Lemli–Opitz syndrome Archived 2010-06-02 at the Wayback Machine
- Pages with script errors
- CS1 maint: url-status
- All articles with unsourced statements
- Articles with unsourced statements from August 2021
- Articles with invalid date parameter in template
- Wikipedia articles incorporating text from the United States National Library of Medicine
- Webarchive template wayback links
- Autosomal recessive disorders
- Cholesterol and steroid metabolism disorders
- Genodermatoses
- Rare diseases
- Syndromes affecting head size
- Syndromes affecting the nervous system
- Syndromes with short stature