Purpura fulminans
| Purpura fulminans | |
|---|---|
| Other names: purpura gangrenosa[1]: 825 | |
Purpura fulminans is an acute, often fatal, thrombotic disorder which manifests as blood spots, bruising and discolouration of the skin resulting from coagulation in small blood vessels within the skin and rapidly leads to skin necrosis and disseminated intravascular coagulation.[2][3]
Signs and symptoms
The presentation of Purpura fulminans is consistent with: hypotension, rash, bleeding from different areas of the body and fever[4]
-
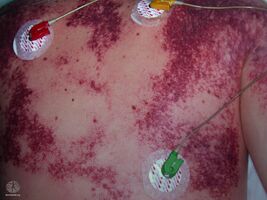
Purpura fulminans
-
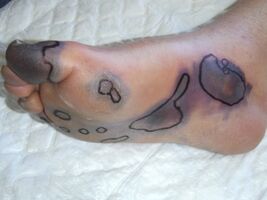
Purpura fulminans
.jpg)
.jpg)
Causes
Purpura fulminans is caused by defects in the protein C anticoagulant pathway. Identification of the cause of purpura fulminans often depends on the patient’s age and circumstances of presentation.[2]
Congenital protein C deficiency
Congenital (inherited) defects in protein C activity are autosomal dominant and may be partial or severe loss of function.[2][5] Hundreds of natural mutations of the protein C gene (PROC) have been identified.[6][7]
Acquired protein C deficiency
Acquired protein C deficiency is caused by either depletion of available protein C in plasma or decreased protein C synthesis (caused by administration of vitamin K antagonists, severe liver failure or complications of prematurity).[8]
Severe acute sepsis
Purpura fulminans is a presenting feature of severe acute sepsis, such as Neisseria meningitidis, Streptococcus pneumoniae, Group A and B Streptococci, and less commonly with Haemophilus influenzae, Staphylococcus aureus, or Plasmodium falciparum (malaria) infections, particularly in individuals with asplenia.[2]
Combination of sepsis and partial congenital defect
In some cases, a combination of sepsis and a partial congenital defect in the protein C anticoagulant pathway initiates purpura fulminans.[9]
Unknown
In rare instances, purpura fulminans is an autoimmune manifestation against protein C or protein S after normally benign infections, such as chicken pox.[2][10] Sometimes purpura fulminans has unknown cause.[2]
Pathophysiology
Regardless of the underlying cause of purpura fulminans, the mechanism of disease is similar with deficiency in protein C concentration or decrease in protein C activity which promotes blood clotting (thrombosis).[11]
In cases of severe sepsis, there is widespread activation of the acute systemic inflammatory response, including activation of the coagulation and complement pathways, as well as endothelial dysfunction. Activated protein C helps regulate the systemic inflammatory response. During sepsis, signalling by the inflammatory cytokines, interleukin-1 and tumour necrosis factor, mediate altered protein transcription in the systemic inflammatory response, resulting in decreased synthesis of the regulatory proteins antithrombin, protein C and protein S, with increased synthesis of prothrombotic proteins Factor VIII, von Willebrand factor, and fibrinogen. Activated protein C binds to endothelial protein C receptor and subsequently cleaves the endothelial cell protease activated receptor-1, not only altering coagulation profiles but down-regulating pro-inflammatory and pro-apoptotic mediators, up-regulation of anti-inflammatory and anti-apoptotic pathways and stabilization of the endothelial cell barrier functions.[12]
Systemic coagulation activation may lead to depletion of circulating coagulation factors and platelets, which subsequently lead to bleeding.[13] In early purpura fulminans, lesion progression correlates with the histological appearance of blockage of small skin blood vessels with blood clots causing capillary dilation and congestion with red blood cells. In later stage lesions, there is irreversible endothelial ischaemic injury with extravasation of blood cells into the dermis and gangrenous necrosis, sometimes with secondary infection.[14]
The depletion of anticoagulant and anti-inflammatory proteins, in particular, protein C and its co-factor, protein S, may also promote thrombus formation, inhibit fibrinolysis and lead to further activation of the inflammatory pathways.[15][16]
Diagnosis
Early purpura fulminans lesions look similar to traumatic skin bleeds or purpuric rashes, such as immune thrombocytopenic purpura or thrombotic thrombocytopenic purpura; however, purpura fulminans will rapidly progress to necrosis whereas other purpuric rashes do not.[2] In most cases, differential diagnoses may be distinguished from purpura fulminans by other clinical and laboratory findings.[2]
The initial appearance of purpura fulminans lesions is of well-demarcated erythematous lesions which progress rapidly to develop irregular central areas of blue-black haemorrhagic necrosis.[2] Advancing areas of necrosis are often surrounded by a thin border of erythema that fades into adjacent unaffected skin. Haemorrhage into the necrotic skin causes purpura fulminans lesions to become painful, dark and raised, sometimes with vesicle or blister (bulla) formation.[17]
The distribution of purpura fulminans lesions may be different according to the underlying pathogenesis.[2] Purpura fulminans in severe sepsis typically develops in the distal extremities and progresses proximally or appears as a generalised or diffuse rash affecting the whole body surface.[17] In cases of severe inheritable protein C deficiency, purpura fulminans with disseminated intravascular coagulation manifests within a few hours or days after birth.[8][18]
Laboratory investigations
The cardinal features of purpura investigations are the same as those of disseminated intravascular coagulation: prolonged plasma clotting times, thrombocytopenia, reduced plasma fibrinogen concentration, increased plasma fibrin-degradation products and occasionally microangiopathic haemolysis.[2]
Prevention
For people who have severe congenital protein C deficiency, protein C replacement therapies are available, which is indicated and approved for use in the United States and Europe for the prevention of purpura fulminans. Protein C replacement is often in combination with anticoagulation therapy of injectable low molecular weight heparin or oral warfarin.[19][20][21][22][23] Before initiating warfarin therapy, a few days of therapeutic heparin may be administered to prevent warfarin necrosis and other progressive or recurrent thrombotic complications.[8][23]
Complications of prevention
The amount of fresh frozen plasma required to reverse disseminated intravascular coagulation associated with purpura fulminans may lead to complications of fluid overload and death, especially in neonates,[8] such as transfusion-related acute lung injury. Exposure to multiple plasma donors over time increases the cumulative risk for transfusion-associated viral infection and allergic reaction to donor proteins found in fresh frozen plasma.[8]
Allergic reactions and alloantibody formation are also potential complications, as with any protein replacement therapy.[8]
Concomitant warfarin therapy in subjects with congenital protein C deficiency is associated with an increased risk of warfarin skin necrosis.[8]
Treatment
Early stage sepsis-associated purpura fulminans may be reversible with quick therapeutic intervention.[2][8] Treatment is mainly removing the underlying cause and degree of clotting abnormalities and with supportive treatment (antibiotics, volume expansion, tissue oxygenation, etc.). Thus, treatment includes aggressive management of the septic state.
Purpura fulminans with disseminated intravascular coagulation should be urgently treated with fresh frozen plasma (10–20 mL/kg every 8–12 hours) and/or protein C concentrate to replace pro-coagulant and anticoagulant plasma proteins that have been depleted by the disseminated intravascular coagulation process.[2][3][5][8]
Protein C in plasma in the steady state has a half life of 6- to 10-hour, therefore, patients with severe protein C deficiency and presenting with purpura fulminans can be treated acutely with an initial bolus of protein C concentrate 100 IU/kg followed by 50 IU /kg every 6 hours.[8] A total of 1 IU/kg of protein C concentrate or 1 mL/kg of fresh frozen plasma will increase the plasma concentration of protein C by 1 IU/dL.[8] Cases with comorbid pathological bleeding may require additional transfusions with platelet concentrate (10–15 mL/kg) or cryoprecipitate (5 mL/kg).[2]
Established soft tissue necrosis may require surgical removal of the dead tissue, fasciotomy, amputation or reconstructive surgery.[2]
Prognosis
Purpura fulminans lesions, once established, often progress within 24 to 48 hours to full-thickness skin necrosis or soft-tissue necrosis. Once purpura fulminans lesions progress to full-thickness skin necrosis, healing takes between 4–8 weeks and leaves large scars.[2]
Without treatment, necrotic soft tissue may become gangrenous, leading to loss of limbs.[2] Purpura fulminans is often accompanied by micro-vascular thrombosis and haemorrhagic infarction in other tissues, such as the lungs, kidneys, central nervous system and adrenal glands, leading to multiple organ failure, and causes initial high mortality and long-term morbidity in survivors. Purpura fulminans may also lead to severe large vessel venous thrombosis if untreated in its early stages.[2]
Purpura fulminans secondary to severe infection is self-limiting.[2] In cases of homozygous protein C deficiency, episodes of purpura fulminans and other thrombotic events are recurrent.[5] Moreover, infant survival due to maintenance replacement therapy is often associated with mental retardation and/or visual impairment.[2][24] For post-infection purpura fulminans, new lesions will occur while neutralising autoantibodies are present (1–2 weeks after presentation).[10]
A multi-disciplinary care team is usually required for rehabilitation after purpura fulminans.[2]
Epidemiology
Purpura fulminans is rare and most commonly occurs in babies and small children[25] but can also be a rare manifestation in adults when it is associated with severe infections.[26] For example, Meningococcal septicaemia is complicated by purpura fulminans in 10–20% of cases among children.[27] Purpura fulminans associated with congenital (inherited) protein C deficiency occurs in 1:500,000–1,000,000 live births.[28]
Research
Due to the rarity of purpura fulminans and its occurrence in vulnerable patient groups like children research on the condition is very limited and evidence based knowledge is scarce. Currently, there is only one purpura fulminans related clinical research project, http://www.sapfire-registry.org/ Archived 2020-12-03 at the Wayback Machine, which is registered with ClinicalTrials.gov.
History
Purpura fulminans was first described by Guelliot in 1884.[29]
References
- ↑ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 0-7216-2921-0.
- ↑ 2.00 2.01 2.02 2.03 2.04 2.05 2.06 2.07 2.08 2.09 2.10 2.11 2.12 2.13 2.14 2.15 2.16 2.17 2.18 2.19 2.20 Chalmers E, Cooper P, Forman K, Grimley C, Khair K, Minford A, Morgan M, Mumford AD (2011). "Purpura fulminans: recognition, diagnosis and management". Arch Dis Child. 96 (11): 1066–1071. doi:10.1136/adc.2010.199919. PMID 21233082. S2CID 206846385.
- ↑ 3.0 3.1 Ghosh SK, Bandyopadhyay D, Dutta A (2009). "Purpura fulminans: a cutaneous marker of disseminated intravascular coagulation". West J Emerg Med. 10 (1): 41. PMC 2672288. PMID 19561767.
- ↑ Perera, Thomas B.; Murphy-Lavoie, Heather M. (2021). "Purpura Fulminans". StatPearls. StatPearls Publishing. Archived from the original on 14 January 2021. Retrieved 22 June 2021.
- ↑ 5.0 5.1 5.2 Estelles A, Garcia-Plaza I, Dasi A, Aznar J, Duart M, Sanz G, Perez-Requejo JL, Espana F, Jimenez C, Abeledo G (1984). "Severe inherited "homozygous" protein C deficiency in a newborn infant". Thromb Haemost. 52 (1): 53–56. doi:10.1055/s-0038-1661136. PMID 6548587.
- ↑ D'Ursi P, Marino F, Caprera A, Milanesi L, Faioni EM, Rovida E (2007). "ProCMD: a database and 3D web resource for protein C mutants". BMC Bioinformatics. 8 (Suppl 1): S11. doi:10.1186/1471-2105-8-s1-s11. PMC 1885840. PMID 17430555.
- ↑ Ursitti JA, Petrich BG, Lee PC, Resneck WG, Ye X, Yang J, Randall WR, Bloch RJ, Wang Y (2007). "Role of an alternatively spliced form of alphaII-spectrin in localization of connexin 43 in cardiomyocytes and regulation by stress-activated protein kinase". J Mol Cell Cardiol. 42 (3): 572–581. doi:10.1016/j.yjmcc.2006.11.018. PMC 1983066. PMID 17276456.
- ↑ 8.00 8.01 8.02 8.03 8.04 8.05 8.06 8.07 8.08 8.09 8.10 Goldenberg NA, Manco-Johnson MJ (2008). "Protein C deficiency". Haemophilia. 14 (6): 1214–1221. doi:10.1111/j.1365-2516.2008.01838.x. PMID 19141162. S2CID 2979452.
- ↑ Gurgey A. (1999). "Clinical manifestations in thrombotic children with factor V Leiden mutation". Pediatr Hematol Oncol. 16 (3): 233–237. doi:10.1080/088800199277281. PMID 10326221.
- ↑ 10.0 10.1 Levin M, Eley BS, Louis J, Cohen H, Young L, Heyderman RS (1995). "Postinfectious purpura fulminans caused by an autoantibody directed against protein S". J Pediatr. 127 (3): 355–63. doi:10.1016/s0022-3476(95)70063-3. PMID 7658262.
- ↑ Adcock DM, Brozna J, Marlar RA (1990). "Proposed classification and pathologic mechanisms of purpura fulminans and skin necrosis". Semin Thromb Hemost. 16 (4): 333–340. doi:10.1055/s-2007-1002686. PMID 2281322.
- ↑ Mosnier LO, Zlokovic BV, Griffin JH (2007). "The cytoprotective protein C pathway". Blood. 109 (8): 3161–3172. doi:10.1182/blood-2006-09-003004. PMID 17110453.
- ↑ Kondaveeti S, Hibberd ML, Booy R, Nadel S, Levin M (1999). "Effect of the Factor V Leiden mutation on the severity of meningococcal disease". Pediatr Infect Dis J. 18 (10): 893–896. doi:10.1097/00006454-199910000-00011. PMID 10530586.
- ↑ Adcock DM, Hicks MJ (1990). "Dermatopathology of skin necrosis associated with purpura fulminans". Semin Thromb Hemost. 16 (4): 283–292. doi:10.1055/s-2007-1002681. PMID 2281318.
- ↑ Fourrier F, Lestavel P, Chopin C, Marey A, Goudemand J, Rime A, Mangalaboyi J (1990). "Meningococcemia and purpura fulminans in adults: acute deficiencies of proteins C and S and early treatment with antithrombin III concentrates". Intensive Care Med. 16 (2): 121–124. doi:10.1007/bf01699858. PMID 2139671. S2CID 10997888.
- ↑ Paramo JA, Perez JL, Serrano M, Rocha E (1990). "Types 1 and 2 plasminogen activator inhibitor and tumor necrosis factor alpha in patients with sepsis". Thromb Haemost. 64 (1): 3–6. doi:10.1055/s-0038-1647143. PMID 2274926.
- ↑ 17.0 17.1 Francis RB (1990). "Acquired purpura fulminans". Semin Thromb Hemost. 16 (4): 310–325. doi:10.1055/s-2007-1002684. PMID 2281320.
- ↑ Marlar RA, Montgomery RR, Broekmans AW (1989). "Diagnosis and treatment of homozygous protein C deficiency. Report of the Working Party on Homozygous Protein C Deficiency of the Subcommittee on Protein C and Protein S, International Committee on Thrombosis and Haemostasis". J Pediatr. 114 (4): 528–534. doi:10.1016/s0022-3476(89)80688-2. PMID 2647943.
- ↑ Dreyfus M, Magny JF, Bridey F, Schwarz HP, Planche C, Dehan M, Tchernia G (1991). "Treatment of homozygous protein C deficiency and neonatal purpura fulminans with a purified protein C concentrate". N Engl J Med. 325 (22): 1565–1568. doi:10.1056/nejm199111283252207. PMID 1944440.
- ↑ Dreyfus M, Masterson M, David M, Rivard GE, Muller FM, Kreuz W, Beeg T, Minford A, Allgrove J, Cohen JD, et al. (1995). "Replacement therapy with a monoclonal antibody purified protein C concentrate in newborns with severe congenital protein C deficiency". Semin Thromb Hemost. 21 (4): 371–381. doi:10.1055/s-2007-1000658. PMID 8747700.
- ↑ "Human protein C: new preparations. Effective replacement therapy for some clotting disorders". Prescrire Int. 12 (63): 11–13. 2003. PMID 12602374.
- ↑ Hartman KR, Manco-Johnson M, Rawlings JS, Bower DJ, Marlar RA (1989). "Homozygous protein C deficiency: early treatment with warfarin". Am J Pediatr Hematol Oncol. 11 (4): 395–401. PMID 2618972.
- ↑ 23.0 23.1 Monagle P, Andrew M, Halton J, Marlar R, Jardine L, Vegh P, Johnston M, Webber C, Massicotte MP (1998). "Homozygous protein C deficiency: description of a new mutation and successful treatment with low molecular weight heparin". Thromb Haemost. 79 (4): 756–761. doi:10.1055/s-0037-1615060. PMID 9569188.
- ↑ Gladson CL, Groncy P, Griffin JH (1987). "Coumarin necrosis, neonatal purpura fulminans, and protein C deficiency". Arch Dermatol. 123 (12): 1701a–1706a. doi:10.1001/archderm.1987.01660360157029. PMID 2961308.
- ↑ Tuddenham, EG, Takase T, Thomas, AE, Awidi AS, Madanat FF, Abu Hajir MM, Kernoff PB Hoffbrand AV (1989). "Homozygous protein C deficiency with delayed onset of symptoms at 7 to 10 months". Thromb Res. 53 (5): 475–84. doi:10.1016/0049-3848(89)90202-8. PMID 2660320.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Lerolle N, Carlotti A, Melican K, Aubey F, Pierrot M, Diehl JL, Caille V, Hekimian G, Gandrille S, Mandet C, Bruneval P, Dumenil G, Borgel D (2013). "Assessment of the interplay between blood and skin vascular abnormalities in adult purpura fulminans". Am J Respir Crit Care Med. 188 (6): 684–692. doi:10.1164/rccm.201302-0228oc. PMID 23924269.
- ↑ Wong VK, Hitchcock W, Mason WH (1989). "Meningococcal infections in children: a review of 100 cases". Pediatr Infect Dis J. 8 (4): 224–227. PMID 2654860.
- ↑ Manco-Johnson MJ, Abshire TC, Jacobson LJ, Marlar RA (1991). "Severe neonatal protein C deficiency: prevalence and thrombotic risk". J Pediatr. 119 (5): 793–798. doi:10.1016/s0022-3476(05)80305-1. PMID 1834822. Archived from the original on 2020-06-30. Retrieved 2020-12-13.
- ↑ Guelliot A (1884). "Note sur trois cas de purpusa infectieux foudroyant". Un Med Sci Nord-Est. 8: 25.
External links
- Medscape: Purpura Fulminans Archived 2021-08-29 at the Wayback Machine
| Classification |
|---|