Pemphigus foliaceus
| Pemphigus foliaceus | |
|---|---|
.jpg) | |
| Pemphigus foliaceus | |
| Specialty | Dermatology[1] |
| Symptoms | Blisters, scale, [1] pain, burning, itch[2] |
| Duration | Many years[2] |
| Causes | Autoimmune,[1] drugs: penicillamine, captopril[2] |
| Risk factors | Genetic, some medications[2] |
| Medication | Dapsone, hydroxychloroquine, corticosteroids (topical or by mouth), topical calcineurin inhibitors[2] |
| Frequency | Adults age 40-50 years,[2][3] males = females[2] |
Pemphigus foliaceus (PF), is an autoimmune disease of the skin.[1] It begins with multiple small fragile blisters that burst as they appear, leading to crust formation on a background of moist damaged skin which bleeds easily.[2] With time, less blisters form and the predominant scaly crusts may give the appearance of 'cornflakes'.[2] Affected people may remain well for many years and may complain of pain, burning and itch.[2] The inside of the mouth is usually not affected.[2]
It is caused by antibodies targeting desmoglein 1 (Dsg-1).[2][3] Drugs that trigger PF include penicillamine and ACE inhibitors, particularly captopril.[2]
Diagnosis is based on appearance, correlated with findings from biopsy of the affected skin, and testing either a blood sample or a skin sample for the antibodies that cause PF.[2] Biopsy samples typically show acantholysis of the upper granular layer of skin.[2] The stratum corneum maybe completely separated from the underlying epidermis or it may be absent altogether.[2] The epidermis, particularly the upper layer, shows widespread intercellular IgG on direct immunofluorescence.[2] Antibodies to Dsg-1 can be detected using ELISA, in people who have confirmed positive indirect immunofluorescence results.[2] Pemphigus vegetans may appear similar and have similar test results.[2] Treatment options include dapsone, hydroxychloroquine, and corticosteroids by mouth or applied to skin.[2] Topical calcineurin inhibitors are another option.[2]
It is usually seen in people aged 40 to 60 years, with an equal distribution between males and females.[2][3] It is more common in Tunisia and Brazil, where around 15% run in families.[2] Pierre Louis Alphée Cazenave gave the first account of the condition in 1844.[4]
Signs and symptoms
The presentation of this condition are consistent with blistering ,erythema and acantholysis[5]
-
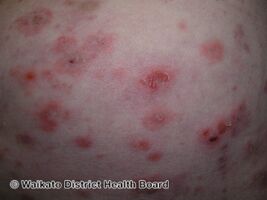
Pemphigus foliaceus
-
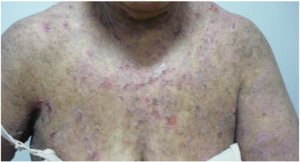
Pemphigus foliaceus
-
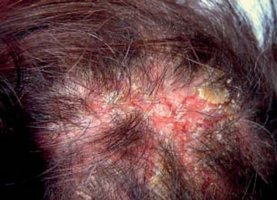
Pemphigus foliaceus scalp
-
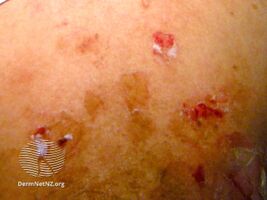
Pemphigus foliaceus(drug induced)
.jpg)


.jpg)
Cause and mechanism
It is caused by (antibodies) targeting desmoglein 1 (Dsg-1).[2] In PF the immune system produces antibodies against desmogleins, proteins in the skin that behave like a glue that keeps skin cells together and the skin intact. Skin cells in PF therefore separate from each other and fluid leaks, forming blisters.[6]
If there is an autoimmune IgG buildup in the epidermis, then nearly all of the antibodies are aimed against Dsg-1. The effect of the antibodies and the immunological pathway is most likely one of three mechanisms:
- Steric hindrance of the desmoglein 1: The antibody caps off the site for intracellular binding to another keratinocyte.
- Activation of an endocytic pathway: The antibody activates a pathway which causes an internalization of desmogleïn 1, which in turn causes a loss of adhesion.
- Disruption of function: In this case, the antibody blocks the desmoglein 1 from being formed into a desmosome. This in turn causes a loss of adhesion with acantholysis as a result.[citation needed]
Diagnosis
Diagnosis is based on appearance, correlated with findings from biopsy of the affected skin, and testing either a blood sample or a skin sample for the antibodies that cause PF.[2] Biopsy samples of affected skin shows acantholysis of the upper granular layer of skin.[2] The stratum corneum maybe completely separated from the underlying epidermis or it may be absent altogether.[2] Long acantholytic cells might be seen clutching to the underside of the stratum corneum.[2] The epidermis, particularly the upper layer, shows widespread intercellular IgG on direct immunofluorescence.[2] Antibodies to Dsg-1 can be detected using ELISA, in people who have confirmed positive indirect immunofluorescence results.[2]
Treatment
It is usually treated with corticosteroids.[7] Other options include dapsone, hydroxychloroquine, and topical calcineurin inhibitors.[2]
Epidemiology
It is usually seen in people aged 50 to 60 years, with an equal distribution between males and females.[3] It is more common in Tunisia and Brazil, where around 15% run in families.[2]
History
Pierre Louis Alphée Cazenave gave the first account of the condition in 1844.[4]
See also
References
- ↑ 1.0 1.1 1.2 1.3 Motta, Adriana; González, Luis Fernando; García, Gonzalo; Guzmán, Jennifer; Prada, Lorena; Herrera, Hugo; Rolon, Mariam (2022). "10. Vesiculobullous Inflammatory Diseases". Atlas of Dermatology: Inflammatory, Infectious and Tumoral Skin Diseases. Springer. pp. 284–285. ISBN 978-3-030-84106-5. Archived from the original on 2022-03-13. Retrieved 2022-03-12.
- ↑ 2.00 2.01 2.02 2.03 2.04 2.05 2.06 2.07 2.08 2.09 2.10 2.11 2.12 2.13 2.14 2.15 2.16 2.17 2.18 2.19 2.20 2.21 2.22 2.23 2.24 2.25 2.26 2.27 2.28 2.29 2.30 2.31 James, William D.; Elston, Dirk; Treat, James R.; Rosenbach, Misha A.; Neuhaus, Isaac (2020). "21. Chronic blistering dermatoses". Andrews' Diseases of the Skin: Clinical Dermatology (13th ed.). Edinburgh: Elsevier. p. 457-458. ISBN 978-0-323-54753-6. Archived from the original on 2022-03-13. Retrieved 2022-03-12.
- ↑ 3.0 3.1 3.2 3.3 Lepe, Kenia; Yarrarapu, Siva Naga S.; Zito, Patrick M. (2021). "Pemphigus Foliaceus". StatPearls. StatPearls Publishing. Archived from the original on 2022-03-13. Retrieved 2022-03-12.
- ↑ 4.0 4.1 Crissey, John Thorne; Parish, Lawrence C.; Holubar, Karl (2013). "The French willanists". Historical Atlas of Dermatology and Dermatologists. CRC Press. p. 60. ISBN 978-1-84184-864-8. Archived from the original on 2023-07-21. Retrieved 2023-07-19.
- ↑ "Pemphigus foliaceus | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 21 March 2021. Retrieved 26 May 2021.
- ↑ NIH Publication No. 15–7083 (June 2015). "Questions and Answers about Pemphigus". National Institute of Arthritis and Musculoskeletal and Skin Disease. NIH. Archived from the original on 17 August 2015. Retrieved 23 October 2015.
- ↑ Porro, Adriana Maria; Hans Filho, Günter; Santi, Claudia Giuli (2019). "Consensus on the treatment of autoimmune bullous dermatoses:pemphigus vulgaris and pemphigus foliaceus - Brazilian Society ofDermatology". Anais Brasileiros de Dermatologia. 94 (2 Suppl 1): 20–32. doi:10.1590/abd1806-4841.2019940206. ISSN 0365-0596. Archived from the original on 28 August 2021. Retrieved 26 May 2021.
External links
| Classification | |
|---|---|
| External resources |