Papular mucinosis
| Papular mucinosis | |
|---|---|
| Specialty | Dermatology |
Papular mucinosis (also known as scleromyxedema,[1][2] "generalized lichen myxedematosus" and "sclerodermoid lichen myxedematosus") is a rare skin disease. Localized and disseminated cases are called papular mucinosis or lichen myxedematosus while generalized, confluent papular forms with sclerosis are called scleromyxedema. Frequently, all three forms are regarded as papular mucinosis. However, some authors restrict it to only mild cases. Another form, acral persistent papular mucinosis is regarded as a separate entity.
Signs and symptoms
Papular mucinosis is chronic and may be progressive. The dermal layer of the skin breaks out into small and solid bumps, usually conical in shape and measured from 2 to 4 mm or sometimes flat-topped papules. Unlike pustules, these bumps do not contain pus. Instead they contain mucin, a waxy substance of mucus, the body's natural and protective lubricant found in saliva and epithelial cells in lungs and the sensitive part of the nose. They usually come in clusters such as linear arrays. Less frequently, urticarial, nodular, or sometimes annular lesions may be appreciated. The dorsal aspect of the hands, face, elbows, and extensor portions of the extremities are most frequently affected. Mucosal lesions are absent. The coalescence of papules on the face, particularly on the glabella, results in longitudinal folding and gives the appearance of a leonine facies.
In scleromyxedema, symptoms can occur on larger part of the body. Redness and scleroderma-like induration occurs on the skin. In addition, the mobility of the lips, hands, arms, and legs is reduced. Proximal myopathy, inflammatory polyarthritis, central nervous system symptoms, esophageal aperistalsis, and hoarseness are among the notable systemic symptoms. If viscera is involved, the disease will be fatal. The dermatoneuro syndrome is a rare neurological complication of the disease presenting with fever, seizures and altered mental status.
Pathology
In the pathology of this dermal condition the dermis contains mucin in abundance and collagen deposition[3]
-
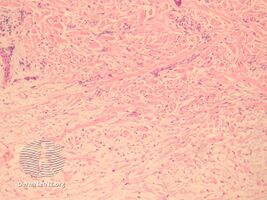
pathology-Papular mucinosis
-
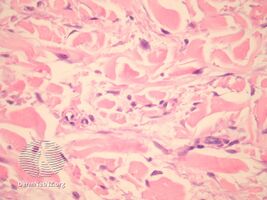
pathology-Papular mucinosis
-
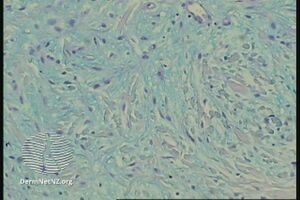
pathology-Papular mucinosis
.jpg)
.jpg)
.jpg)
Diagnosis
Classification
Papular mucinosis may be divided into several types or variants:[1]
Treatment
There is no effective treatment for this condition. It has been reported that clearance of lesions can be done with melphalan and cyclophosphamide alone or in combination with prednisone. Both isotretinoin and etretinate have also been shown to improve the conditions. All medications listed can cause adverse symptoms, with isotretinoin and etretinate particularly dangerous since they are both teratogens. Other attempted treatments include interferon-alpha, cyclosporine, PUVA photochemotherapy, electron-beam therapy, IVIg and dermabrasion. However, the overall prognosis for the disease is poor. There are reported instances of remission of the disease when treated with a combination of Revlimid and Dexamethasone over a 24-month period.[citation needed]
Incidence
Papular mucinosis affects adults of both sexes equally and appears between ages 30 and 80. Recently,[when?] it has been reported in patients infected with the HIV/AIDS virus.[citation needed] It has also been associated with certain paraproteinemias such as IgG lambda paraproteinemia.[citation needed]
See also
Notes
- ↑ 1.0 1.1 Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 1-4160-2999-0.
- ↑ Applebaum DS, Daulat S, Duvic M (August 2014). "Clinicopathological Challenge: Multiple Skin-Colored Papules With Diffuse Sclerosis". JAMA Dermatol. 150 (8): 893–894. doi:10.1001/jamadermatol.2014.14. Archived from the original on December 29, 2012. Retrieved August 18, 2014.
- ↑ "Papular mucinosis pathology | DermNet NZ". www.dermnetnz.org. Archived from the original on 2 March 2021. Retrieved 3 July 2021.
References
- "What is Papular mucinosis?", The Star (Malaysia), December 20, 2006.
- "Papular Mucinosis" Archived 2012-02-05 at the Wayback Machine, Dermatology Online Journal Vol. 7 No. 2.
External links
| Classification | |
|---|---|
| External resources |