PMS2




Mismatch repair endonuclease PMS2 is an enzyme that in humans is encoded by the PMS2 gene.[4]
Function
This gene is one of the PMS2 gene family members which are found in clusters on chromosome 7. Human PMS2 related genes are located at bands 7p12, 7p13, 7q11, and 7q22. Exons 1 through 5 of these homologues share high degree of identity to human PMS2 [5] The product of this gene is involved in DNA mismatch repair. The protein forms a heterodimer with MLH1 and this complex interacts with MSH2 bound to mismatched bases. Defects in this gene are associated with hereditary nonpolyposis colorectal cancer, with Turcot syndrome, and are a cause of supratentorial primitive neuroectodermal tumors. Alternatively spliced transcript variants have been observed.[6]
Mismatch repair and endonuclease activity
PMS2 is involved in mismatch repair and is known to have latent endonuclease activity that depends on the integrity of the meta-binding motif in MutL homologs. As an endonuclease, PMS2 introduces nicks into a discontinuous DNA strand.[7]
Interactions
PMS2 has been shown to interact with MLH1 by forming the heterodimer MutLα.[8][9][10][11][12][13] There is competition between MLH3, PMS1, and PMS2 for the interacting domain on MLH1, which is located in residues 492-742.[9]
The interacting domains in PMS2 have heptad repeats that are characteristic of leucine zipper proteins.[9] MLH1 interacts with PMS2 at residues 506-756.[10]
The MutS heterodimers, MutSα and MutSβ, associate with MutLα upon mismatch binding. MutLα is believed to join the mismatch recognition step to other processes, including: removal of mismatches from the new DNA strand, resynthesis of the degraded DNA, and repair of the nick in the DNA.[13] MutLα is shown to have weak ATPase activity and also possesses endonuclease activity that introduces nicks into the discontinuous strand of DNA. This facilitates 5' to 3' degradation of the mismatched DNA strand by EXO1.[13] The active site of MutLα is located on the PMS2 subunit. PMS1 and PMS2 compete for interaction with MLH1.[13] Proteins in the interactome of PMS2 have been identified by tandem affinity purification.[13][14]
Human PMS2 is expressed at very low levels and is not believed to be strongly cell cycle regulated.[15]
Interactions involving p53 and p73
PMS2 has also been shown to interact with p53 and p73. In the absence of p53, PMS2-deficient and PMS2-proficient cells are still capable of arresting the cell cycle at the G2/M checkpoint when treated with cisplatin.[16] Cells that are deficient in p53 and PMS2, exhibit increased sensitivity to anticancer agents. PMS2 is a protective mediator of cell survival in p53-deficient cells and modulates protective DNA damage response pathways independently of p53.[16] PMS2 and MLH1 can protect cells from cell death by counteracting p73-mediated apoptosis in a mismatch repair dependent manner.[16]
PMS2 can interact with p73 to enhance cisplatin-induced apoptosis by stabilizing p73. Cisplatin stimulates the interaction between PMS2 and p73, which is dependent on c-Abl.[12] The MutLα complex may function as an adapter to bring p73 to the site of damaged DNA and also act as an activator of p73, due to the presence of PMS2.[12] It may also be possibly for overexpressed PMS2 to stimulate apoptosis in the absence of MLH1 and in the presence of p73 and cisplatin due to the stabilizing actions of PMS2 on p73.[12] Upon DNA damage, p53 induces cell cycle arrest through the p21/WAF pathway and initiates repair by expression of MLH1 and PMS2.[11] The MSH1/PMS2 complex acts as a sensor of the extent of the damage to the DNA, and initiates apoptosis by stabilizing p73 if the damage is beyond repair.[11] Loss of PMS2 does not always lead to instability of MLH1 since it can also form complexes with MLH3 and PMS1.[17]
Clinical significance
Mutations
PMS2 is a gene that encodes for DNA repair proteins involved in mismatch repair. The PMS2 gene is located on chromosome 7p22 and it consists of 15 exons. Exon 11 of the PMS2 gene has a coding repeat of eight adenosines.[18]
Comprehensive genomic profiling of 100,000 human cancer samples revealed that mutations in the promoter region of PMS2 are significantly associated with high tumor mutational burden (TMB), particularly in melanoma.[19] TMB has been shown to be a reliable predictor of whether a patient may respond to cancer immunotherapy, where high TMB is associated with more favorable treatment outcomes.[20]
Heterozygous germline mutations in DNA mismatch repair genes like PMS2 lead to autosomal dominant Lynch syndrome. Only 2% of families that have Lynch syndrome have mutations in the PMS2 gene.[21] The age of patients when they first presented with PMS2-associated Lynch syndrome varies greatly, with a reported range of 23 to 77 years.[22]
In rare cases, a homozygous defect may cause this syndrome. In such cases a child inherits the gene mutation from both parents and the condition is called Turcot syndrome or Constitutional MMR Deficiency (CMMR-D).[23] Up until 2011, 36 patients with brain tumors due to biallelic PMS2 germline mutations have been reported.[23] Inheritance of Turcot syndrome can be dominant or recessive. Recessive inheritance of Turcot syndrome is caused by compound heterozygous mutations in PMS2.[24] 31 out of 57 families reported with CMMR-D have germline PMS2 mutations.[25] 19 out of 60 PMS2 homozygous or compound heterozygous mutation carriers had gastrointestinal cancer or adenomas as the first manifestation of CMMR-D.[25] Presence of pseudogenes can cause confusion when identifying mutations in PMS2, leading to false positive conclusions of the presence of mutated PMS2.[18]
Deficiency and overexpression
Overexpression of PMS2 results in hypermutability and DNA damage tolerance.[26] Deficiency of PMS2 also contributes to genetic instability by allowing for mutations to propagate due to reduced MMR function.[26] It has been shown that PMS2-/- mice developed lymphomas and sarcomas. It was also shown that male mice that are PMS2-/- are sterile, indicating that PMS2 may have a role in spermatogenesis.[7]
Role in normal colon
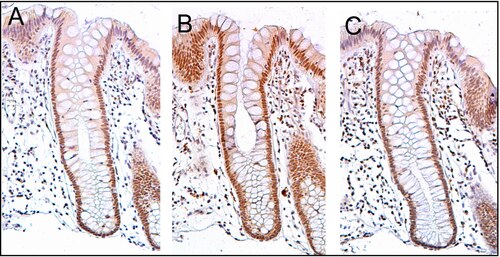
PMS2 is usually expressed at a high level in cell nuclei of enterocytes (absorptive cells) within the colonic crypts lining the inner surface of the colon (see image, panel A). DNA repair, involving high expression of PMS2, ERCC1 and ERCC4 (XPF) proteins, appears to be very active in colon crypts in normal, non-neoplastic colonic epithelium. In the case of PMS2, the expression level in normal colonic epithelium is high in 77% to 100% of crypts.[27]
Cells are produced at the crypt base and migrate upward along the crypt axis before being shed into the colonic lumen days later.[28] There are 5 to 6 stem cells at the bases of the crypts.[28] If the stem cells at the base of the crypt express PMS2, generally all several thousand cells of the crypt[29] will also express PMS2. This is indicated by the brown color seen by immunostaining of PMS2 in most of the enterocytes in the crypt in panel A of the image in this section. Similar expression of ERCC4 (XPF) and ERCC1 occurs in the thousands of enterocytes in each colonic crypt of the normal colonic epithelium.
The tissue section in the image shown here was also counterstained with hematoxylin to stain DNA in nuclei a blue-gray color. Nuclei of cells in the lamina propria (cells which are below and surround the epithelial crypts) largely show hematoxylin blue-gray color and have little expression of PMS2, ERCC1 or ERCC4 (XPF).
Colon cancer
About 88% of cells of epithelial origin in colon cancers, and about 50% of the colon crypts in the epithelium within 10 cm adjacent to cancers (in the field defects from which the cancers likely arose) have reduced or absent expression of PMS2.[27]
Deficiencies in PMS2 in colon epithelium appear to mostly be due to epigenetic repression. In tumors classified as mismatch repair deficient and lacking, in a majority PMS2 expression is deficient because of lack of its pairing partner MLH1.[30] Pairing of PMS2 with MLH1 stabilizes.[31] The loss of MLH1 in sporadic cancers was due to epigenetic silencing caused by promoter methylation in 65 out of 66 cases. In 16 cancers Pms2 was deficient even though MLH1 protein expression was present. Of these 16 cases, no cause was determined for 10, but 6 were found to have a heterozygous germline mutation in Pms2, followed by likely loss of heterozygosity in the tumor. Thus only 6 of 119 tumors lacking expression for Pms2 (5%) were due to mutation of PMS2.
Coordination with ERCC1 and ERCC4 (XPF)

When PMS2 is reduced in colonic crypts in a field defect, it is most often associated with reduced expression of DNA repair enzymes ERCC1 and ERCC4 (XPF) as well (see images in this section). A deficiency in ERCC1 and/or ERCC4 (XPF) would cause DNA damage accumulation. Such excess DNA damage often leads to apoptosis.[32] However, an added defect in PMS2 can inhibit this apoptosis.[33][34] Thus, an added deficiency in PMS2 likely would be selected for in the face of the increased DNA damages when ERCC1 and/or ERCC4 (XPF) are deficient. When ERCC1 deficient Chinese hamster ovary cells were repeatedly subjected to DNA damage, of five clones derived from the surviving cells, three were mutated in Pms2.[35]
Progression to colon cancer
ERCC1, PMS2 double mutant Chinese hamster ovary cells, when exposed to Ultraviolet light (a DNA damaging agent), showed a 7,375-fold greater mutation frequency than wild type Chinese hamster ovary cells, and a 967-fold greater mutation frequency than the cells defective in ERCC1, alone.[35] Thus colonic cell deficiency in both ERCC1 and PMS2 causes genome instability. A similar genetically unstable situation is expected for cells doubly defective for PMS2 and ERCC4 (XPF). This instability would likely enhance progression to colon cancer by causing a mutator phenotype,[36] and account for the presence of the cells doubly deficient in PMS2 and ERCC1 [or PMS2 and ERCC4 (XPF)] in field defects associated with colon cancer. As indicated by Harper and Elledge,[37] defects in the ability to properly respond to and repair DNA damage underlie many forms of cancer.
References
- ^ a b c GRCh38: Ensembl release 89: ENSG00000122512 - Ensembl, May 2017
- ^ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ Nicolaides NC, Papadopoulos N, Liu B, Wei YF, Carter KC, Ruben SM, Rosen CA, Haseltine WA, Fleischmann RD, Fraser CM (Sep 1994). "Mutations of two PMS homologues in hereditary nonpolyposis colon cancer". Nature. 371 (6492): 75–80. Bibcode:1994Natur.371...75N. doi:10.1038/371075a0. PMID 8072530. S2CID 4244907.
- ^ Nicolaides NC, Carter KC, Shell BK, Papadopoulos N, Vogelstein B, Kinzler KW (November 1995). "Genomic organization of the human PMS2 gene family". Genomics. 30 (2): 195–206. doi:10.1006/geno.1995.9885. PMID 8586419.
- ^ "Entrez Gene: PMS2 PMS2 postmeiotic segregation increased 2 (S. cerevisiae)".
- ^ a b van Oers JM, Roa S, Werling U, Liu Y, Genschel J, Hou H, Sellers RS, Modrich P, Scharff MD, Edelmann W (12 July 2010). "PMS2 endonuclease activity has distinct biological functions and is essential for genome maintenance". Proc. Natl. Acad. Sci. U.S.A. 107 (30): 13384–9. Bibcode:2010PNAS..10713384V. doi:10.1073/pnas.1008589107. PMC 2922181. PMID 20624957.
- ^ Mac Partlin M, Homer E, Robinson H, McCormick CJ, Crouch DH, Durant ST, Matheson EC, Hall AG, Gillespie DA, Brown R (February 2003). "Interactions of the DNA mismatch repair proteins MLH1 and MSH2 with c-MYC and MAX". Oncogene. 22 (6): 819–25. doi:10.1038/sj.onc.1206252. PMID 12584560.
- ^ a b c Kondo E, Horii A, Fukushige S (April 2001). "The interacting domains of three MutL heterodimers in man: hMLH1 interacts with 36 homologous amino acid residues within hMLH3, hPMS1 and hPMS2". Nucleic Acids Res. 29 (8): 1695–702. doi:10.1093/nar/29.8.1695. PMC 31313. PMID 11292842.
- ^ a b Guerrette S, Acharya S, Fishel R (March 1999). "The interaction of the human MutL homologues in hereditary nonpolyposis colon cancer". J. Biol. Chem. 274 (10): 6336–41. doi:10.1074/jbc.274.10.6336. PMID 10037723.
- ^ a b c Chen J, Sadowski I (March 2005). "Identification of the mismatch repair genes PMS2 and MLH1 as p53 target genes by using serial analysis of binding elements". Proc. Natl. Acad. Sci. U.S.A. 102 (13): 4813–8. Bibcode:2005PNAS..102.4813C. doi:10.1073/pnas.0407069102. PMC 555698. PMID 15781865.
- ^ a b c d Shimodaira H, Yoshioka-Yamashita A, Kolodner RD, Wang JY (March 2003). "Interaction of mismatch repair protein PMS2 and the p53-related transcription factor p73 in apoptosis response to cisplatin". Proc. Natl. Acad. Sci. U.S.A. 100 (5): 2420–5. Bibcode:2003PNAS..100.2420S. doi:10.1073/pnas.0438031100. PMC 151356. PMID 12601175.
- ^ "PMS2 Gene". The GeneCards Human Gene Database. Weizmann Institute of Science.
- ^ Meyers M, Theodosiou M, Acharya S, Odegaard E, Wilson T, Lewis JE, Davis TW, Wilson-Van Patten C, Fishel R, Boothman DA (January 1997). "Cell cycle regulation of the human DNA mismatch repair genes hMSH2, hMLH1, and hPMS2". Cancer Res. 57 (2): 206–8. PMID 9000555.
- ^ a b c Fedier A, Ruefenacht UB, Schwarz VA, Haller U, Fink D (October 2002). "Increased sensitivity of p53-deficient cells to anticancer agents due to loss of Pms2". Br. J. Cancer. 87 (9): 1027–33. doi:10.1038/sj.bjc.6600599. PMC 2364320. PMID 12434296.
- ^ Nakagawa H, Lockman JC, Frankel WL, Hampel H, Steenblock K, Burgart LJ, Thibodeau SN, de la Chapelle A (July 2004). "Mismatch repair gene PMS2: disease-causing germline mutations are frequent in patients whose tumors stain negative for PMS2 protein, but paralogous genes obscure mutation detection and interpretation". Cancer Res. 64 (14): 4721–7. doi:10.1158/0008-5472.CAN-03-2879. PMID 15256438.
- ^ a b Chadwick RB, Meek JE, Prior TW, Peltomaki P, de La Chapelle A (December 2000). "Polymorphisms in a pseudogene highly homologous to PMS2". Hum. Mutat. 16 (6): 530. doi:10.1002/1098-1004(200012)16:6<530::AID-HUMU15>3.0.CO;2-6. PMID 11102987. S2CID 23159181.
- ^ Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, Schrock A, Campbell B, Shlien A, Chmielecki J, Huang F, He Y, Sun J, Tabori U, Kennedy M, Lieber DS, Roels S, White J, Otto GA, Ross JS, Garraway L, Miller VA, Stephens PJ, Frampton GM (April 2017). "Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden". Genome Med. 9 (34): epub. doi:10.1186/s13073-017-0424-2. PMC 5395719. PMID 28420421.>
- ^ Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, Stephens PJ, Daniels GA, Kurzrock R (November 2017). "Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers". Mol. Cancer Ther. 16 (11): 2598–2608. doi:10.1158/1535-7163.MCT-17-0386. PMC 5670009. PMID 28835386.>
- ^ "PMS2 - PMS2 postmeiotic segregation increased 2 (S. cerevisiae)". Genetics Home Reference. U.S. National Library of Medicine.
- ^ Senter L, Clendenning M, Sotamaa K, Hampel H, Green J, Potter JD, Lindblom A, Lagerstedt K, Thibodeau SN, Lindor NM, Young J, Winship I, Dowty JG, White DM, Hopper JL, Baglietto L, Jenkins MA, de la Chapelle A (August 2008). "The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations". Gastroenterology. 135 (2): 419–28. doi:10.1053/j.gastro.2008.04.026. PMC 2759321. PMID 18602922.
- ^ a b Johannesma PC, van der Klift HM, van Grieken NC, Troost D, Te Riele H, Jacobs MA, Postma TJ, Heideman DA, Tops CM, Wijnen JT, Menko FH (September 2011). "Childhood brain tumours due to germline bi-allelic mismatch repair gene mutations". Clin. Genet. 80 (3): 243–55. doi:10.1111/j.1399-0004.2011.01635.x. PMID 21261604. S2CID 23927730.
- ^ De Rosa M, Fasano C, Panariello L, Scarano MI, Belli G, Iannelli A, Ciciliano F, Izzo P (March 2000). "Evidence for a recessive inheritance of Turcot's syndrome caused by compound heterozygous mutations within the PMS2 gene". Oncogene. 19 (13): 1719–1723. doi:10.1038/sj.onc.1203447. PMID 10763829.
- ^ a b Herkert JC, Niessen RC, Olderode-Berends MJ, Veenstra-Knol HE, Vos YJ, van der Klift HM, Scheenstra R, Tops CM, Karrenbeld A, Peters FT, Hofstra RM, Kleibeuker JH, Sijmons RH (May 2011). "Paediatric intestinal cancer and polyposis due to bi-allelic PMS2 mutations: case series, review and follow-up guidelines". Eur. J. Cancer. 47 (7): 965–82. doi:10.1016/j.ejca.2011.01.013. PMID 21376568.
- ^ a b Gibson SL, Narayanan L, Hegan DC, Buermeyer AB, Liskay RM, Glazer PM (December 2006). "Overexpression of the DNA mismatch repair factor, PMS2, confers hypermutability and DNA damage tolerance". Cancer Lett. 244 (2): 195–202. doi:10.1016/j.canlet.2005.12.009. PMID 16426742.
- ^ a b c d e Facista A, Nguyen H, Lewis C, Prasad AR, Ramsey L, Zaitlin B, Nfonsam V, Krouse RS, Bernstein H, Payne CM, Stern S, Oatman N, Banerjee B, Bernstein C (2012). "Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer". Genome Integr. 3 (1): 3. doi:10.1186/2041-9414-3-3. PMC 3351028. PMID 22494821.
- ^ a b Baker AM, Cereser B, Melton S, Fletcher AG, Rodriguez-Justo M, Tadrous PJ, Humphries A, Elia G, McDonald SA, Wright NA, Simons BD, Jansen M, Graham TA (2014). "Quantification of crypt and stem cell evolution in the normal and neoplastic human colon". Cell Rep. 8 (4): 940–7. doi:10.1016/j.celrep.2014.07.019. PMC 4471679. PMID 25127143.
- ^ Nooteboom M, Johnson R, Taylor RW, Wright NA, Lightowlers RN, Kirkwood TB, Mathers JC, Turnbull DM, Greaves LC (2010). "Age-associated mitochondrial DNA mutations lead to small but significant changes in cell proliferation and apoptosis in human colonic crypts". Aging Cell. 9 (1): 96–9. doi:10.1111/j.1474-9726.2009.00531.x. PMC 2816353. PMID 19878146.
- ^ Truninger K, Menigatti M, Luz J, Russell A, Haider R, Gebbers JO, Bannwart F, Yurtsever H, Neuweiler J, Riehle HM, Cattaruzza MS, Heinimann K, Schär P, Jiricny J, Marra G (2005). "Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer". Gastroenterology. 128 (5): 1160–71. doi:10.1053/j.gastro.2005.01.056. PMID 15887099.
- ^ Chang DK, Ricciardiello L, Goel A, Chang CL, Boland CR (2000). "Steady-state regulation of the human DNA mismatch repair system". J. Biol. Chem. 275 (24): 18424–31. doi:10.1074/jbc.M001140200. PMID 10747992.
- ^ Norbury CJ, Zhivotovsky B (2004). "DNA damage-induced apoptosis". Oncogene. 23 (16): 2797–808. doi:10.1038/sj.onc.1207532. PMID 15077143.
- ^ Fukuhara S, Chang I, Mitsui Y, Chiyomaru T, Yamamura S, Majid S, Saini S, Deng G, Gill A, Wong DK, Shiina H, Nonomura N, Lau YF, Dahiya R, Tanaka Y (2015). "Functional role of DNA mismatch repair gene PMS2 in prostate cancer cells". Oncotarget. 6 (18): 16341–51. doi:10.18632/oncotarget.3854. PMC 4599273. PMID 26036629.
- ^ Marinovic-Terzic I, Yoshioka-Yamashita A, Shimodaira H, Avdievich E, Hunton IC, Kolodner RD, Edelmann W, Wang JY (2008). "Apoptotic function of human PMS2 compromised by the nonsynonymous single-nucleotide polymorphic variant R20Q". Proc. Natl. Acad. Sci. U.S.A. 105 (37): 13993–8. Bibcode:2008PNAS..10513993M. doi:10.1073/pnas.0806435105. PMC 2528866. PMID 18768816.
- ^ a b Nara K, Nagashima F, Yasui A (2001). "Highly elevated ultraviolet-induced mutation frequency in isolated Chinese hamster cell lines defective in nucleotide excision repair and mismatch repair proteins". Cancer Res. 61 (1): 50–2. PMID 11196196.
- ^ Loeb LA (2011). "Human cancers express mutator phenotypes: origin, consequences and targeting". Nat. Rev. Cancer. 11 (6): 450–7. doi:10.1038/nrc3063. PMC 4007007. PMID 21593786.
- ^ Harper JW, Elledge SJ (2007). "The DNA damage response: ten years after". Mol. Cell. 28 (5): 739–45. doi:10.1016/j.molcel.2007.11.015. PMID 18082599.
External links
- FAQs on HNPCC Archived 2007-08-15 at the Wayback Machine from the National Institute of Health
- GeneReviews/NCBI/NIH/UW entry on Lynch syndrome
PDB gallery | |
|---|---|
|


