Osteochondroma
| Osteochondroma | |
|---|---|
| Other names: Osteocartilaginous exostoses (not recommended),[1] ecchondroma, cartilaginous exostosis[2] | |
 | |
| X-ray knee (side view): ossification in the peritendinous tissues in a person with osteochondroma. | |
| Specialty | Orthopedics[3] |
| Diagnostic method | Medical imaging[4] |
| Frequency | Common, around 35% of non-cancerous and 8% of all bone tumors, M>F[1] |
Osteochondroma is a type of non-cancerous bone tumor belonging to the group of cartilaginous tumors.[5] They tend to be a single tumor in a long bone of the arm or leg.[6]
The tumors take the form of cartilage-capped bony projections or outgrowth on the surface of bones exostoses.[7][8] It is characterized as a type of overgrowth that can occur in any bone where cartilage forms bone.[9][7] Additionally, flat bones such as the pelvis and scapula (shoulder blade) may be affected.[10] Hereditary multiple exostoses usually present during childhood. Yet, the vast majority of affected individuals become clinically manifest by the time they reach adolescence.[7][11]
Osteochondromas do not result from injury and the exact cause remains unknown. Recent research has indicated that multiple osteochondromas is an autosomal dominant inherited disease. Germ line mutations in EXT1 and EXT2 genes located on chromosomes 8 and 11 have been associated with the cause of the disease.[12]
The treatment choice for osteochondroma is surgical removal of solitary lesion or partial excision of the outgrowth, when symptoms cause motion limitations or nerve and blood vessel impingements.[8][13] In hereditary multiple exostoses the indications of surgery are based upon multiple factors that are taken collectively, namely: patient's age, tumor location and number, accompanying symptomatology, esthetic concerns, family history and underlying gene mutation.[9][7] A variety of surgical procedures have been employed to remedy hereditary multiple exostoses such as osteochondroma excision, bone lengthening, corrective osteotomy and hemiepiphysiodesis. Sometimes a combination of the previous procedures is used.[7] The indicators of surgical success in regard to disease and patient characteristics are greatly disputable.[7] Because most studies of hereditary multiple exostoses are retrospective and of limited sample size with missing data, the best evidence for each of the currently practiced surgical procedures is lacking.[7]
Osteochondromas occur in 3% of the general population and represent 35% of all benign tumors and 8% of all bone tumors. The majority of these tumors are solitary non-hereditary lesions and approximately 15% of osteochondromas occur as hereditary multiple exostoses preferably known as hereditary multiple osteochondromas (HMOs).[8][13]
Signs and symptoms
Limited normal functions and movements are caused by osteochondromas growing slowly and inwardly. The majority of osteochondromas are symptomless and are found incidentally. Each individual with osteochondroma may experience symptoms differently and most of the time individuals will experience no symptoms at all. Some of the most common symptoms are a hard immobile painless palpable mass, adjacent muscle soreness, and pressure or irritation with heavy exercising.[10] Major symptoms arise when complications such as fractures, bone deformity or mechanical joint problems occur. If the occurrence of an osteochondroma is near a nerve or a blood vessel, the affected limb can experience numbness, weakness, loss of pulse or color change. Periodic changes in the blood flow can also take place. Approximately 20% of patients experiencing nerve compression commonly acknowledge vascular compression, arterial thrombosis, aneurysm, and pseudoaneurysm. Formation of pseudoaneurysm and venous thrombosis lead to claudication, pain, acute ischemia, and symptoms of phlebitis. If the tumor is found under a tendon, it can cause pain during movement causing restriction of joint motion. Pain can also occur due to bursal inflammation, swelling or fracture at the base of the tumor stalk. Some of the clinical signs and symptoms of malignant osteochondroma are pain, swelling, and mass enlargement.[8]
Mechanism
Osteochondromas are long and slender, pedunculated on a stalk often taking the shape of a cauliflower. The cartilage cap is covered by fibrous perichondrium and continues with the periosteum of the underlying bone. The cartilage cap is less than 2 cm thick and the thickness decreases with age. A cap more than 2 cm thick, indicates malignant transformation of a tumor. The cartilage cap merges with the epiphyseal area of the long bones called spongiosa. In the spongiosa, the chondrocytes are arranged in accordance with the epiphyseal growth plate. The spongiosa of the stalk continues with the underlying cancellous bone. Fractures within the stalk causes fibroblastic proliferation and formation of a new bone. Development of bursa takes place over the osteochondroma, which is attached to the perichondrium of the cap. Inflammation of the bone is indicated by the bursal wall lined by the synovium. As a result, patients may have swelling for years related to the location and site of the lesion indicative of mechanical obstruction, nerve impingement, pseudoaneurysm of the overlying vessel, fracture at the stalk of the lesion, or formation of bursa over the osteochondroma.[13] Heparan sulphate (HS) are glycosaminoglycans which are involved in the formation of proteoglycans. The biosynthesis of HS takes place in the Golgi apparatus and endoplasmic reticulum, where glycosaminoglycans chains are maintained by type II glycosyltransferases encoded by EXOSTOSIN genes EXT1 and EXT2. Decreased levels of HS leads to mutations in EXT1 or EXT2 causing skeletal abnormality.[14] The underlying mechanism for solitary and multiple osteochondromas have been associated with genetic alterations in EXT1 or EXT2 genes located on chromosomes 8 and 11. Approximately 65% of osteochondromas arise in the EXT1 gene loci on chromosome 8 and 35% arise in EXT2 gene loci on chromosome 11. About 70–75% of multiple osteochondromas are caused by point mutations, often involving deletion of single or multiple axons as found in 10% of all hereditary cases. In about 10–15% of all cases no genomic alterations are detected. The mechanism behind the formation of multiple osteochondroma is large genomic deletions of EXT1 and EXT2 genes. The identified mechanism behind solitary osteochondromas is the homozygous deletions of the EXT1 gene.[11] However, the exact cause of osteochondroma is unknown.[10] Additionally, the molecular basis of genetics and clinical variability of multiple osteochondroma as well as the underlying causes for the malignant transformation and the onset of osteochondroma in EXT negative patients is also currently unknown.[15]
Diagnosis
Osteochondromas are often asymptomatic and may not cause any kind of discomfort. They are often found accidentally when an X-ray is done for an unrelated reason.[16]
- X-rays are the first tests performed that characterize a lesion. They show a clear picture of dense structures of bones, and will also indicate bone growth pertaining to osteochondroma.[10][16]
- Computed tomography (CT) scan can identify the bony lesion in great details and show the presence of calcification. These tests also provide great details, especially in soft tissues with the aide of cross-sectional images.
- Magnetic resonance imaging (MRI) is the most accurate method for detecting bone masses in symptomatic cases to depict precise morphology of a tumor. It is used to verify if the palpable mass is continuous with the cortex of the affected bone and to differentiate an osteochondroma from other lesions on the surface of the bone. MRI can also be used to look for cartilage on the surface of tumor and can depict any vascular complications caused by the tumor. An MRI can identify tumors of the spinal column and is often used to diagnose low grade osteosarcoma.[8][16]
- Ultrasound is done if aneurysms or pseudoaneurysms and venous or arterial thrombosis is suspected. Ultrasound is an accurate method for examining the cartilaginous cap of the osteochondroma. It is also a way of pinpointing bursitis. However, it cannot be used to predict if the growth of tumor is inward in regards to the cap.[8]
- Angiography is used to detect vascular lesions caused by osteochondroma due to ossified cartilaginous cap. It is also used to characterize malignant transformation lesions through neovascularity.[8]
- Clinical testing such as sequence analysis can be done of the entire coding regions of both EXT1 and EXT2 to detect mutations.[17]
- A biopsy of the tissue sample of the tumor can also be taken to check for cancer.[16]
Tests for osteochondroma can also identify diseases such as secondary peripheral chondrosarcoma and multiple osteochondromatosis. In large, secondary chondrosarcoma arises at the site of osteochondroma due to increased thickness of the cartilage cap indicating potential malignant transformation. The symptoms of multiple osteochondromatosis are similar to solitary osteochondroma, but they are often more severe. Painless bumps can arise at the site of tumor and pain and other discomforts can also take place if pressure is put on the soft tissues, nerves, or blood vessels.[8][16] Dysplasia Epiphysealis Hemimelica (DEH) or Trevor's disease and metachondromatosis (MC) are considered differential diagnosis of both solitary and hereditary osteochondromas. DEH is described as a type of over growth at one or more epiphyses. Similar to osteochondroma, DEH is diagnosed prior to 15 years of age and the growth of lesions end at puberty, when the growth plates close. Metachondromatosis is a rare disorder that exhibit symptoms of both multiple osteochondromas and enchondromas in children and is also inherited in autosomal dominant mode.[18]
A type that contains fat is known as an osteolipochondroma (osteo, bone, lipos, fat, + chondros, cartilage, oma, tumor).[19]
-
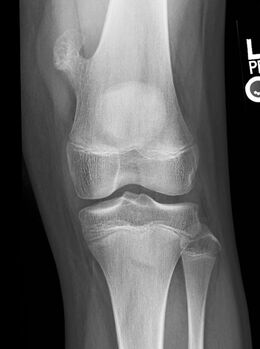
Osteochondroma arising from the thigh bone, near the knee
-

Osteochondroma arising from the large long bone of lower leg, near the knee
-

Osteochondroma arising from long bone of the upper arm, near shoulder



Treatment

Osteochondromas are benign lesions and do not affect life expectancy.[18] Complete excision of osteochondroma is curative and the reoccurrences take place when the removal of tumor is incomplete. Multiple reoccurrences in a well-excised lesion indicate that it may be malignant.[8] The risk of malignant transformation takes place in 1–5% of individuals.[18] If any symptoms of cancerous tumor takes place, then the patient should be evaluated by a bone specialist.[16] No treatment is necessary for Solitary osteochondromas that are asymptomatic. Treatments for solitary osteochondroma are careful observation over time and taking regular x-rays to monitor any changes in the tumor.[16] If the lesion is causing pain with activity, nerve or vessel impingement, or if the bone growth has fully matured and the presence of a large cartilage cap is prominent, then it is advised that the tumor be surgically removed.[20][21]
Osteochondromas have a low rate of malignancy (<1%) and resection of the tumor is suggested if symptoms such as pain, limitation of movement, or impingement on nerves or vessels occur. Resection of the tumor also takes place when the tumor increases in size and progresses towards malignancy. During surgical resection, the entire lesion along with the cartilaginous cap should be removed to minimize any chances of reoccurrences.[13] Surgical treatment becomes the sole treatment of choice if common complications such as fractures, symptoms of peripheral nerves such as paresthesia, paraplegia, peroneal neuropathy, and upper limb neuropathy take place. A prophylactic resection is suggested if the lesion lies next to a vessel.
Depending on the size and location of the tumor, the time it takes to return to normal daily activities varies between individuals. Limitation on some activities is advised if pain or discomfort persists after surgical excision.[16]
Epidemiology
Osteochondroma is one of the most common non-cancerous bone tumors.[1] The average age at diagnosis is 18 years, and males are affected more than females.[1] They constitute around 35% of non-cancerous and 8% of all bone tumors, but there maybe many more that go undetected. About 15% comprise multiple lesions.[1]
Research
Research done using Zebrafish dackel (dak) have shown that in EXT2-/- Zebrafish, chondrocytes fail to undergo terminal differentiation and bone formation fails to progress from pre-osteoblasts stage to osteoblasts. Instead, abnormal lipid deposition and premature adipocyte differentiation takes place. The expression of xbp1, master regulator of osterix gets reduced, suggesting that unfolded proteins responses may play a role in pathogenesis of multiple osteochondroma. The research concludes that heparan sulphates are required for terminal differentiation and formation of scaffold that is needed for bone development. At least one copy of EXT2 gene is needed for proper bone development and to maintain the balance between bone and fat cell lineages. Due to homozygous loss of EXT2 function, leads to imbalance between cartilage, bone, and fat cell lineages. These observations in null zebrafish points toward the musculoskeletal defects observed in patients with multiple osteochondroma. Due to the findings of bone-fat imbalance in Zebra fish model, future studies should address status of lipid composition in patients with multiple osteochondroma.[14] Research conducted using sequencing methods has identified a novel frame shift mutation at the glycosyltransferase domain (c.1457insG) located at codon 486 of exon 6 of the EXT1 gene, that causes multiple osteochondromas. This study was conducted in two multiple osteochondroma (MO) patients from the Chinese descent (same family) and the results were validated with four other members of the same MO family and 200 unrelated healthy subjects. The results of the mutations were validated using two different sequencing methods (Exome and Sanger). The results of immunohistochemistry and multiple sequence alignment supports the cause of MO being a mutation in EXT1 gene. However, the exact molecular mechanism of multiple osteochondroma remains unclear. The EXT1 gene encodes the endoplasmic reticulum-resident type II transmembrane glycosyltransferase, which catalyzes polymerization of heparin sulfate chain at the endoplasmic reticulum and the Golgi apparatus. Heparin sulfate regulates signal transduction during chondrocyte differentiation, ossification, and apoptosis. Malfunction in heparin sulfate synthesis causes chondrocytes to rapidly differentiate. Based on these results future studies should elucidate the underlying molecular mechanism of the glycosyltransferase domain of the EXT1 and its involvement in the development of multiple osteochondromas.[12] Osteochondromas are associated with secondary peripheral chondrosarcomas, but the pathogenesis of the malignant bone tumor remains unknown. Research has demonstrated that chondrocytes with dysfunctional EXT1 is present in solitary osteochondromas, but the EXT1 is functional in sporadic (solitary) secondary peripheral chondrosarcomas. Research indicates that osteochondromas creates a special niche in which wild type cells are mixed in with EXT functional cells. Then these EXT functional cells undergo other mutations, that give rise to secondary peripheral chondrosarcoma, indicating the involvement of an alternative mechanism for the pathogenesis of secondary peripheral chondrosarcoma. Future studies should address the contributing gene that causes the formation of peripheral chondrosarcoma. It should also illustrate what causes chondrocytes functional with EXT1 and EXT2 within the osteochondroma to become more susceptible to mutations leading to malignancy.[11]
References
- ↑ 1.0 1.1 1.2 1.3 1.4 WHO Classification of Tumours Editorial Board, ed. (2020). "Osteochondroma". Soft Tissue and Bone Tumours: WHO Classification of Tumours. Vol. 3 (5th ed.). Lyon (France): International Agency for Research on Cancer. pp. 356–358. ISBN 978-92-832-4503-2. Archived from the original on 2021-06-13. Retrieved 2021-05-10.
- ↑ "ICD-11 - ICD-11 for Mortality and Morbidity Statistics". icd.who.int. Archived from the original on 1 August 2018. Retrieved 6 July 2021.
- ↑ Maruthainar, Nimalan; Bhumbra, Rej; Cannon, Steve (2018). "7. Orthopaedic oncology". In Ramachandran, Manoj (ed.). Basic Orthopaedic Sciences (2nd ed.). CRC Press. p. 118. ISBN 978-1-4441-2098-1. Archived from the original on 2021-06-13. Retrieved 2021-05-02.
- ↑ Bocklage, Therese J.; Quinn, Robert; Verschraegen, Claire; Schmit, Berndt (2014). "16. Cartilaginous tumours of bones and joints". Bone and Soft Tissue Tumors: A Multidisciplinary Review with Case Presentations. London: JP Medical Ltd. pp. 366–409. ISBN 978-1-907816-22-2. Archived from the original on 2021-07-09. Retrieved 2021-08-21.
- ↑ WHO Classification of Tumours Editorial Board, ed. (2020). "Bone tumors". Soft Tissue and Bone Tumours: WHO Classification of Tumours. Vol. 3 (5th ed.). Lyon (France): International Agency for Research on Cancer. p. 338. ISBN 978-92-832-4503-2. Archived from the original on 2021-06-13. Retrieved 2021-05-10.
- ↑ Suster, David; Hung, Yin Pun; Nielsen, G. Petur (1 January 2020). "Differential Diagnosis of Cartilaginous Lesions of Bone". Archives of Pathology & Laboratory Medicine. 144 (1): 71–82. doi:10.5858/arpa.2019-0441-RA. ISSN 0003-9985. Archived from the original on 19 July 2021. Retrieved 17 July 2021.
- ↑ 7.0 7.1 7.2 7.3 7.4 7.5 7.6 EL-Sobky, TA; Samir, S; Atiyya, AN; Mahmoud, S; Aly, AS; Soliman, R (21 March 2018). "Current paediatric orthopaedic practice in hereditary multiple osteochondromas of the forearm: a systematic review". Sicot-J. 4: 10. doi:10.1051/sicotj/2018002. PMC 5863686. PMID 29565244.
- ↑ 8.0 8.1 8.2 8.3 8.4 8.5 8.6 8.7 8.8 Panagiotis, Kitsoulis; Vassiliki Galani; Kalliopi Stefanaki; Georgios Paraskevas; Georgios Karatzias; Niki John Agnantis; Maria Bai (October 2008). "Osteochondromas: Review of the Clinical, Radiological and Pathological Features". In Vivo. 22 (5): 633–646. PMID 18853760. Archived from the original on 11 August 2019. Retrieved 22 March 2014.
- ↑ 9.0 9.1 Wuyts, W; Schmale, GA; Chansky, HA; et al. (21 November 2013). "Hereditary Multiple Osteochondromas". GeneReviews. Archived from the original on 28 January 2017. Retrieved 30 March 2018.
- ↑ 10.0 10.1 10.2 10.3 "Osteochondroma". University of Rochester Medical Center. Archived from the original on 27 May 2020. Retrieved 15 March 2014.
- ↑ 11.0 11.1 11.2 de Andrea, CE; Reijnders CM; Kroon HM; De Jong D; Hogendoorn PC; Szuhai K; Bovée JV (1 March 2012). "Secondary peripheral chondrosarcoma evolving from osteochondroma as a result of outgrowth of cells with functional EXT". Oncogene. 31 (9): 1095–1104. doi:10.1038/onc.2011.311. PMID 21804604. S2CID 11418240.
- ↑ 12.0 12.1 Krahe, Ralf; Zhang, Feng; Liang, Jinlong; Guo, Xiong; Zhang, Yingang; Wen, Yan; Li, Qiang; Zhang, Zengtie; Ma, Weijuan; Dai, Lanlan; Liu, Xuanzhu; Yang, Ling; Wang, Jun (2013). "Exome Sequencing and Functional Analysis Identifies a Novel Mutation in EXT1 Gene That Causes Multiple Osteochondromas". PLOS ONE. 8 (8): e72316. doi:10.1371/journal.pone.0072316. ISSN 1932-6203. PMC 3757002. PMID 24009674.
- ↑ 13.0 13.1 13.2 13.3 Reijnders, Christianne; Liesbeth Hameetman; Judith VMG Bovée (September 2008). "Bone: Osteochondroma". Atlas of Genetics and Cytogenetics in Oncology and Haematology. Archived from the original on 23 November 2019. Retrieved 15 March 2014.
- ↑ 14.0 14.1 Wiweger, Malgorzata; De Andrea CE; Scheepstra KW; Zhao Z; Hogendoorn PC (March 2014). "Possible effects of EXT2 on mesenchymal differentiation– lessons from the zebrafish". Orphanet Journal of Rare Diseases. 9 (35): 35. doi:10.1186/1750-1172-9-35. PMC 4004154. PMID 24628984.
- ↑ Zuntini, M; Salvatore M; Pedrini E; Parra A; Sgariglia F; Magrelli A; Taruscio D; Sangiorgi L (December 2010). "MicroRNA profiling of multiple osteochondromas: identification of disease-specific and normal cartilage signatures". Clin. Genet. 78 (6): 507–516. doi:10.1111/j.1399-0004.2010.01490.x. PMID 20662852. S2CID 21999710.
- ↑ 16.0 16.1 16.2 16.3 16.4 16.5 16.6 16.7 "Osteochondroma". American Academy of Orthopedic Surgeons. Archived from the original on 7 November 2017. Retrieved 15 March 2014.
- ↑ Wuyts, Wim (21 November 2013). GeneReviews. Seattle, WA: University of Washington, Seattle. Archived from the original on 28 January 2017. Retrieved 7 September 2017.
- ↑ 18.0 18.1 18.2 Bovée, Judith VMG (13 February 2008). "Multiple Osteochondromas". Orphanet Journal of Rare Diseases. 3 (1): 3. doi:10.1186/1750-1172-3-3. PMC 2276198. PMID 18271966.
- ↑ MORRIS, CHRISTOPHER W.; Morris, Christopher G.; Press, Academic (1992). Academic Press Dictionary of Science and Technology. Gulf Professional Publishing. p. 1539. ISBN 9780122004001. Archived from the original on 2021-08-28. Retrieved 2020-09-20.
- ↑ "Osteochondroma". Bone Tumor.org. Archived from the original on 25 February 2019. Retrieved 25 March 2014.
- ↑ "Osteochondroma". PhysioPedia. Archived from the original on 9 September 2019. Retrieved 25 March 2014.
External links
- Humpath #2790 Archived 2009-04-13 at the Wayback Machine (Pathology images)
- American Academy of Orthopedic Surgeons Archived 2017-11-07 at the Wayback Machine
| Classification | |
|---|---|
| External resources |