Medullary thyroid cancer
| Medullary thyroid cancer | |
|---|---|
| Other names: MTC | |
 | |
| Micrograph of medullary thyroid carcinoma with amyloid deposition (left of image). Near normal thyroid follicles are also seen (right of image). H&E stain. | |
| Specialty | ENT surgery |
Medullary thyroid cancer is a form of thyroid carcinoma which originates from the parafollicular cells (C cells), which produce the hormone calcitonin.[1] Medullary tumors are the third most common of all thyroid cancers and together make up about 3% of all thyroid cancer cases.[2] MTC was first characterized in 1959.[3]
Approximately 25% of medullary thyroid cancer cases are genetic in nature, caused by a mutation in the RET proto-oncogene. When MTC occurs by itself it is termed sporadic medullary thyroid cancer (SMTC). Medullary thyroid cancer is seen in people with multiple endocrine neoplasia type 2A and 2B. When medullary thyroid cancer occurs without other endocrine tumours it is termed familial medullary thyroid cancer (FMTC).
Signs and symptoms
The major clinical symptom of metastatic medullary thyroid carcinoma is diarrhea; occasionally a patient will have flushing episodes. Both occur particularly with liver metastasis, and either symptom may be the first manifestation of the disease. The flushing that occurs in medullary thyroid carcinoma is indistinguishable from that associated with carcinoid syndrome. In MTC, the flushing, diarrhea, and itching (pruritus) are all caused by elevated levels of calcitonin gene products (calcitonin or calcitonin gene-related peptide).[4] By comparison, the flushing and diarrhea observed in carcinoid syndrome is caused by elevated levels of circulating serotonin.
Medullary thyroid carcinoma may also produce a thyroid nodule and enlarged cervical lymph nodes.[4]
Sites of spread of medullary thyroid carcinoma include local lymph nodes in the neck, lymph nodes in the central portion of the chest (mediastinum), liver, lung, and bone. Spread to other sites such as skin or brain occurs but is uncommon.
Genetics
Mutations (DNA changes) in the RET proto-oncogene, located on chromosome 10, lead to the expression of a mutated receptor tyrosine kinase protein, termed RET (REarranged during Transfection). RET is involved in the regulation of cell growth and development and its germline mutation is responsible for nearly all cases of hereditary or familial medullary thyroid carcinoma. Its germline mutation may also be responsible for the development of hyperparathyroidism and pheochromocytoma. Hereditary medullary thyroid cancer is inherited as an autosomal dominant trait, meaning that each child of an affected parent has a 50% probability of inheriting the mutant RET proto-oncogene from the affected parent. DNA analysis makes it possible to identify children who carry the mutant gene; surgical removal of the thyroid in children who carry the mutant gene is curative if the entire thyroid gland is removed at an early age, before there is spread of the tumor. The parathyroid tumors and pheochromocytomas are removed when they cause clinical symptomatology. Hereditary medullary thyroid carcinoma or multiple endocrine neoplasia (MEN2) accounts for approximately 25% of all medullary thyroid carcinomas.
Seventy-five percent of medullary thyroid carcinoma occurs in individuals without an identifiable family history and is assigned the term "sporadic". Individuals who develop sporadic medullary thyroid carcinoma tend to be older and have more extensive disease at the time of initial presentation than those with a family history (screening is likely to be initiated at an early age in the hereditary form). Approximately 25-60% of sporadic medullary thyroid carcinomas have a somatic mutation (one that occurs within a single "parafollicular" cell) of the RET proto-oncogene. This mutation is presumed to be the initiating event, although there could be other as yet unidentified causes.
Markers
While the increased serum concentration of calcitonin is not harmful, it is useful as a marker which can be tested in blood.[5]
A second marker, carcinoembryonic antigen (CEA), also produced by medullary thyroid carcinoma, is released into the blood and it is useful as a serum or blood tumor marker. In general, measurement of serum CEA is less sensitive than serum calcitonin for detecting the presence of a tumor, but has less minute to minute variability and is therefore useful as an indicator of tumor mass.
Diagnosis
-
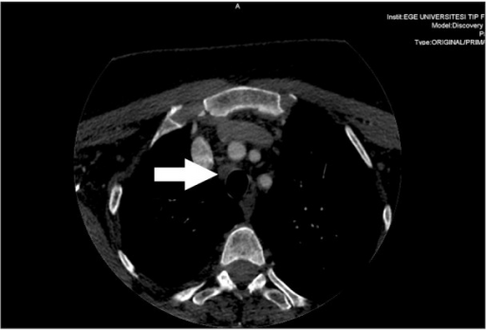
Metastatic central lymph node of a medullary thyroid cancer case
-

Medullary thyroid carcinoma on ultrasound with typical small calcifications (arrows)
.png)

Diagnosis is primarily performed via fine needle aspiration of the lesion of the thyroid to distinguish it from other types of thyroid lesions.[6] Microscopic examination will show an amyloid stroma with hyperplasia of parafollicular cells.
Treatment
Surgery and radiation therapy have been the major treatments for medullary thyroid carcinoma. A plasma level of metanephrines should be checked before surgical thyroidectomy takes place to evaluate for the presence of pheochromocytoma since 25% of people found to have medullary thyroid cancer have the inherited form from the MEN2A syndrome. Undiagnosed pheochromocytoma leads to a very high intraoperative risk of hypertensive crisis and, potentially, death.
Surgery

A total thyroidectomy with bilateral neck dissection is the gold standard for treating medullary thyroid cancer, and is the most definitive means of achieving a cure in patients without distant metastases or extensive nodal involvement. Risks of surgery include loss of vocal control, irreparable nerve damage, death or the need for second operation to clean out residual diseased lymph nodes left behind if the sentinel node biopsy was positive for cancerous spread. Extensive surgery can be effective when the condition is detected early, but a risk for recurrence remains, particularly in patients with multiple positive lymph nodes or extracapsular invasion.[1][7] About half of patients have metastasis to regional lymph nodes at the time of diagnosis.[4]
The European Society of Endocrine Surgeons has published recommendations for managing this condition in gene carriers.[8] The timing of surgery depends on the type of mutation present. For those in the highest risk group, surgery is recommended in the first year of life. In lower risk cases surgery may be delayed up to the age of ten years, the precise timing depending on the mutation and other factors.
Radiation
External beam radiotherapy is recommended when there is a high risk of regional recurrence, even after optimum surgical treatment.[9][10] In this study, patients treated with external beam radiation were compared to a control group. Disease control with radiation was far superior in the group receiving radiation. The authors of the study [14] wrote: "in 40 high risk patients (microscopic residual disease, extraglandular invasion, or lymph node involvement), the local/regional relapse free rate was 86% at 10 years with postoperative external beam radiation (25 patients), and 52% for those with no postoperative external radiation (p = 0.049). To optimize local/regional tumor control, we therefore continue to advise external beam radiation in patients at high risk of local/regional relapse."
Unlike other differentiated thyroid carcinoma, there is no role for radioiodine treatment in medullary-type disease.[11]
Protein kinase inhibitors
Clinical trials of protein kinase inhibitors,[12] which block the abnormal kinase proteins involved in the development and growth of medullary cancer cells, showed clear evidence of response in 10-30% of patients. In the majority of responders there has been less than a 30% decrease in tumor mass, yet the responses have been durable; responses have been stable for periods exceeding 3 years. The major side effects of this class of drug include hypertension, nausea, diarrhea, some cardiac electrical abnormalities, and thrombotic or bleeding episodes.
Vandetanib, trade name Caprelsa, was the first drug (April 2011) to be approved by US Food and Drug Administration (FDA) for treatment of late-stage (metastatic) medullary thyroid cancer in adult patients who are ineligible for surgery.[13]
Cabozantinib, trade name Cometriq, was granted marketing approval (November 2012) by the U.S. FDA for this indication.[14] Cabozantinib which is a potent inhibitor of RET, MET and VEGF was evaluated in a double-blind placebo controlled trial. It was shown to improve overall survival by 5 months for the treated cohort vs. placebo, which was not statistically significant. However, cabozantinib was particularly effective in patients with the RET M918T mutation, extending overall survival by roughly 2 years, doubling survival vs. untreated patient (4 years vs. 2 year). Treatment with cabozantinib did require many dose reduction to mitigate side effects. It has been suggested that the trial dose of 140 mg was excessive, particularly in lower body mass patients. Ongoing trials have been scheduled to identify more optimal dosing regimes. Activity has been observed, in practice at doses of 1.2 mg/kg.
Prognosis
Depending on source, the overall 5-year survival rate for medullary thyroid cancer is 80%,[15] 83%[16] or 86%,[6] and the 10-year survival rate is 75%.[15]
By overall cancer staging into stages I to IV, the 5-year survival rate is 100% at stage I, 98% at stage II, 81% at stage III and 28% at stage IV.[17] The prognosis of MTC is poorer than that of follicular and papillary thyroid cancer when it has metastasized (spread) beyond the thyroid gland.
The prognostic value of measuring calcitonin and carcinoembryonic antigen (CEA) concentrations in the blood was studied in 65 MTC patients who had abnormal calcitonin levels after surgery (total thyroidectomy and lymph node dissection).[18] The prognosis correlated with the rate at which the postoperative calcitonin concentration doubles, termed the calcitonin doubling time (CDT), rather than the pre- or postoperative absolute calcitonin level:
- CDT less than 6 months: 3 patients out of 12 (25%) survived 5 years. 1 patient out of 12 (8%) survived 10 years. All died within 6 months to 13.3 years.
- CDT between 6 months and 2 years: 11 patients out of 12 (92%) survived 5 years. 3 patients out of 8 (37%) survived 10 years. 4 patients out of 12 (25%) survived to the end of the study.
- CDT more than 2 years: 41 patients out of 41 (100%) were alive at the end of the study. These included 1 patient whose calcitonin was stable, and 11 patients who had decreasing calcitonin levels.
The calcitonin doubling time was a better predictor of MTC survival than CEA[18] but following both tests is recommended.[9][19]
References
- ↑ 1.0 1.1 Hu MI, Vassilopoulou-Sellin R, Lustig R, Lamont JP. "Thyroid and Parathyroid Cancers" Archived 2010-02-28 at the Wayback Machine in Pazdur R, Wagman LD, Camphausen KA, Hoskins WJ (Eds) Cancer Management: A Multidisciplinary Approach Archived 2013-10-04 at the Wayback Machine. 11 ed. 2008.
- ↑ Stamatakos M, Paraskeva P, Stefanaki C, Katsaronis P, Lazaris A, Safioleas K, Kontzoglou K (January 2011). "Medullary thyroid carcinoma: The third most common thyroid cancer reviewed". Oncol Lett. 2 (1): 49–53. doi:10.3892/ol.2010.223. PMC 3412473. PMID 22870127.
- ↑ Dionigi G, Bianchi V, Rovera F, et al. (2007). "Medullary thyroid carcinoma: surgical treatment advances". Expert Rev Anticancer Ther. 7 (6): 877–85. doi:10.1586/14737140.7.6.877. PMID 17555398.
- ↑ 4.0 4.1 4.2 Goldman, Lee (2011). Goldman's Cecil Medicine (24th ed.). Philadelphia: Elsevier Saunders. pp. e76. ISBN 978-1437727883.
- ↑ Fragu P (2007). "Calcitonin's fantastic voyage: from hormone to marker of a genetic disorder". Gesnerus. 64 (1–2): 69–92. PMID 17982960.
- ↑ 6.0 6.1 National Cancer Institute > Medullary Thyroid Cancer Archived 2012-06-15 at the Wayback Machine Last Modified: 12/22/2010
- ↑ Schlumberger M, Carlomagno F, Baudin E, Bidart JM, Santoro M (2008). "New therapeutic approaches to treat medullary thyroid carcinoma". Nat Clin Pract Endocrinol Metab. 4 (1): 22–32. doi:10.1038/ncpendmet0717. PMID 18084343.
- ↑ Niederle B, Sebag F, Brauckhoff M (2013) Timing and extent of thyroid surgery for gene carriers of hereditary C cell disease-a consensus statement of the European Society of Endocrine Surgeons (ESES).Langenbecks Arch Surg
- ↑ 9.0 9.1 Thyroid Carcinoma. NCCN guidelines. http://www.nccn.org/professionals/physician_gls/pdf/thyroid.pdf Archived 2021-08-28 at the Wayback Machine
- ↑ Brierley J, Tsang R, Simpson WJ, Gospodarowicz M, Sutcliffe S, Panzarella T (1996). "Medullary thyroid cancer: analyses of survival and prognostic factors and the role of radiation therapy in local control". Thyroid. 6 (4): 305–10. doi:10.1089/thy.1996.6.305. PMID 8875751.
- ↑ Quayle FJ, Moley JF (2005). "Medullary thyroid carcinoma: including MEN 2A and MEN 2B syndromes". J Surg Oncol. 89 (3): 122–9. doi:10.1002/jso.20184. PMID 15719378.
- ↑ "American Thyroid Association - Thyroid Clinical Trials". Archived from the original on 2007-12-12. Retrieved 2007-12-21.
- ↑ "FDA approves new treatment for rare form of thyroid cancer". Archived from the original on 10 April 2011. Retrieved 7 April 2011.
- ↑ "FDA approves Cometriq to treat rare type of thyroid cancer". Archived from the original on 7 July 2014. Retrieved 29 November 2012.
- ↑ 15.0 15.1 Numbers from National Cancer Database in the US, from Page 10 Archived 2016-05-13 at the Wayback Machine in: F. Grünwald; Biersack, H. J.; Grںunwald, F. (2005). Thyroid cancer. Berlin: Springer. ISBN 978-3-540-22309-2.
- ↑ Barbet, J.; Campion, L.; Kraeber-Bodere, F.; Chatal, J. -F.; Group, T. G. T. E. S. (2005). "Prognostic Impact of Serum Calcitonin and Carcinoembryonic Antigen Doubling-Times in Patients with Medullary Thyroid Carcinoma". Journal of Clinical Endocrinology & Metabolism. 90 (11): 6077–6084. doi:10.1210/jc.2005-0044. PMID 16091497.
- ↑ cancer.org > Thyroid Cancer Archived October 18, 2013, at the Wayback Machine By the American Cancer Society. In turn citing: AJCC Cancer Staging Manual (7th ed).
- ↑ 18.0 18.1 Barbet J, Campion L, Kraeber-Bodéré F, Chatal JF (2005). "Prognostic impact of serum calcitonin and carcinoembryonic antigen doubling-times in patients with medullary thyroid carcinoma". J. Clin. Endocrinol. Metab. 90 (11): 6077–84. doi:10.1210/jc.2005-0044. PMID 16091497. Archived from the original on 2022-06-14. Retrieved 2021-04-26.
- ↑ ASCO SEP 3rd edition
External links
| Classification | |
|---|---|
| External resources |