Langerhans cell histiocytosis
| Langerhans cell histiocytosis | |
|---|---|
| Other names: Histiocytosis X[1] | |
 | |
| Micrograph showing a Langerhans cell histiocytosis with the characteristic reniform Langerhans cells accompanied by abundant eosinophils. H&E stain. | |
| Specialty | Hematology |
| Symptoms | Lesions in various tissues, fever, weight loss, diabetes insipidus, tiredness[1] |
| Types | Unifocal, chronic multifocal, multifocal multisystem, Hashimoto-Pritzker disease[2] |
| Diagnostic method | Tissue biopsy, medical imaging[2] |
| Treatment | None, surgery, radiation therapy, chemotherapy[2] |
| Prognosis | Variable[2] |
| Frequency | 1 to 2 per 100,000[3] |
Langerhans cell histiocytosis (LCH) is an abnormal increase in Langerhans immune cells from the bone marrow, which than migrate to the skin and other organs.[4] They can form a type of tumor, known as a granuloma.[4][5] Symptoms may include lesions in various tissues such as skin or bone, fever, weight loss, diabetes insipidus, or tiredness.[1]
LCH is due to a mutation in the MAPKinase pathway.[1] It is part of a group of syndromes called histiocytoses, which are characterized by an increased number of histiocytes (an old term for dendritic cells).[1][4] Diagnosis is confirmed by medical imaging and biopsy.[2] Blood tests may show low red blood cells, and occasionally low white blood cells and low platelets.[1] Many consider it a form of cancer, but there is still some disagreement.[2][6]
Mild cases may simple resolve on their own without treatment.[2] Otherwise treatment options may include some combination of surgery, radiation therapy, or chemotherapy.[2] LCH affects about 1 to 2 people per 100,000.[3] Most cases start in childhood;[3] though adults may also be affected.[4] Males are more commonly affected than females.[1] Hispanic people are more commonly affected.[1]
Evidence of the disease has been found in mummy dating back to 900-790. B.C.[7] LCH was previously known as "histiocytosis X".[8] Subtype include chronic multifocal LCH, previously known as "Hand–Schüller–Christian disease", unifocal LCH, previously known as "eosinophilic granuloma", multifocal multisystem LCH, previously known as "Letterer-Siwe disease", and Hashimoto-Pritzker disease.[2][9]
Signs and symptoms
LCH provokes a non-specific inflammatory response, which includes fever, lethargy, and weight loss.
Either one or several parts of the body can be involved, with clinical features depending on which organ is affected, resulting in an array of signs and symptoms from mild to life-threatening.[1]
- Bone involvement can cause bone pain and swelling. The skull is most frequently affected, followed by long bones of arms and legs. Lytic bone lesions can lead to fractures.[6] Signs and symptoms due to bone damage reflect the type of bone affected, such as ear infections with temporal bone involvement, and spinal cord damage with vertebral damage.[1] Disease of the jaw may result in swollen and bleeding gums and teeth problems.[1]
- Bone marrow involvement can lead to a low blood count, with low plateletes being prominent.[1]
- Liver and spleen involvement causes enlargement of these organs, in addition to low protein, edema, jaundice and large lymph glands.[1][4] Bleeding easily and anemia may be worse if the spleen enlarges.[1]
- Endocrine system can be affected when the disease involves the hypothalamic pituitary axis.[10][11] Anterior pituitary hormone deficiency is usually permanent.[12] Involvement of the pituitary stalk may result in diabetes insipidus, with excessive thirst and passing urine frequently. If the front of the pituitary gland is affected, abnormalities in thyroid, growth, adrenal and some sexual hormones may occur.[1][13]
- Nervous system involvement such as of the cerebellum can result in difficulty walking, hand tremor and difficulty speaking.[1]
- Lungs and gastrointestinal tract involvement may not always result in symptoms and be diagnosed incidentally because of lung nodules on radiographs.[14] Difficulty breathing and coughing blood can arise from lung involvement and bloody diarrhoea can result from lesions in the intestinal tract.[1]
-
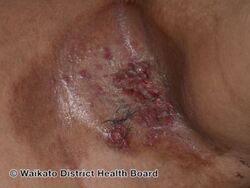
Langerhans histiocytosis
-
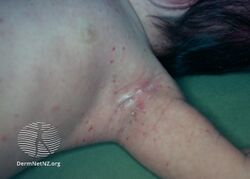
Histiocytosis (dermal infiltrative langerhans)
-
![Langerhans cell histiocytosis[15]](/media/thumb/1/11/LCH-2.jpg/250px-LCH-2.jpg)
Langerhans cell histiocytosis[15]
-
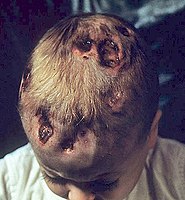
A person with chronic multifocal Langerhans cell histiocytosis, a subtype of LCH.
-
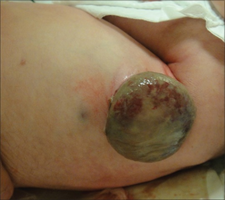
large bump on tummy of newborn with Langerhans cell histiocytosis
.jpg)
.jpg)
![Langerhans cell histiocytosis[15]](/wiki/File:LCH-2.jpg)


Causes
LCH is due to mutations in the MAPKinase pathway.[1] It is part of a group of syndromes called histiocytoses, which are characterized by an abnormal proliferation of histiocytes (an archaic term for activated dendritic cells and macrophages), which were also termed Langerhans cells. These diseases are related to other forms of abnormal proliferation of white blood cells, such as leukemias and lymphomas.[1]
LCH is usually a sporadic and non-hereditary condition but familial clustering has been noted in limited number of cases. Hashimoto-Pritzker disease is a congenital self-healing variant of Hand-Schüller-Christian disease.[16]
Describing LCH as a cancer has caused some controversy.[6] Some sources such as the National Cancer Institute describe it as a type of cancer,[17] while other sources specifically state it is not a type of cancer.[18]
Pathophysiology
The pathogenesis of Langerhans cell histiocytosis (LCH) is a matter of debate. There are ongoing investigations to determine whether LCH is a reactive (non-cancerous) or neoplastic (cancerous) process. Arguments supporting the reactive nature of LCH include the occurrence of spontaneous remissions, the extensive secretion of multiple cytokines by dendritic cells and bystander-cells (a phenomenon known as cytokine storm) in the lesional tissue, favorable prognosis and relatively good survival rate in patients without organ dysfunction or risk organ involvement.[19][20]
On the other hand, the infiltration of organs by monoclonal population of pathologic cells, and the successful treatment of subset of disseminated disease using chemotherapeutic regimens are all consistent with a neoplastic process.[21][22][23] In addition, a demonstration, using X chromosome–linked DNA probes, of LCH as a monoclonal proliferation provided additional support for the neoplastic origin of this disease.[24] While clonality is an important attribute of cancer, its presence does not prove that a proliferative process is neoplastic. Recurrent cytogenetic or genomic abnormalities would also be required to demonstrate convincingly that LCH is a malignancy.
An activating somatic mutation of a proto-oncogene in the Raf family, the BRAF gene, was detected in 35 of 61 (57%) LCH biopsy samples with mutations being more common in patients younger than 10 years (76%) than in patients aged 10 years and older (44%).[25] This study documented the first recurrent mutation in LCH samples. Two independent studies have confirmed this finding.[26][27] Presence of this activating mutation could support the notion to characterize LCH as myeloproliferative disorder.
Diagnosis


Diagnosis is confirmed histologically by tissue biopsy. Hematoxylin-eosin stain of biopsy slide will show features of Langerhans Cell e.g. distinct cell margin, pink granular cytoplasm. Presence of Birbeck granules on electron microscopy and immuno-cytochemical features e. g. CD1 positivity are more specific. Initially routine blood tests e.g. full blood count, liver function test, U&Es, bone profile are done to determine disease extent and rule out other causes.[citation needed]
Imaging may be evident in chest X-rays with micronodular and reticular changes of the lungs with cyst formation in advanced cases. MRI and High-resolution CT may show small, cavitated nodules with thin-walled cysts. MRI scan of the brain can show three groups of lesions such as tumourous/granulomatous lesions, nontumourous/granulomatous lesions, and atrophy. Tumourous lesions are usually found in the hypothalamic-pituitary axis with space-occupying lesions with or without calcifications. In non-tumourous lesions, there is a symmetrical hyperintense T2 signal with hypointense or hyperintense T1 signal extending from grey matter into the white matter. In the basal ganglia, MRI shows a hyperintense T1 signal in the globus pallidus.[28]
Assessment of endocrine function and bonemarrow biopsy are also performed when indicated.[citation needed]
- S-100 protein is expressed in a cytoplasmic pattern[29][30]
- peanut agglutinin (PNA) is expressed on the cell surface and perinuclearly[31][32]
- major histocompatibility (MHC) class II is expressed (because histiocytes are macrophages)
- CD1a[29]
- langerin (CD207), a Langerhans Cell–restricted protein that induces the formation of Birbeck granules and is constitutively associated with them, is a highly specific marker.[33][34]
Classification
| Alternative names |
|---|
| Histiocytosis X
Histiocytosis X syndrome |
| Subordinate terms |
|
Histiocytosis X, unspecified
Disseminated Langerhans cell histiocytosis (clinical) |
The disease spectrum results from clonal accumulation and proliferation of cells resembling the epidermal dendritic cells called Langerhans cells, sometimes called dendritic cell histiocytosis. These cells in combination with lymphocytes, eosinophils, and normal histiocytes form typical LCH lesions that can be found in almost any organ.[36] A similar set of diseases has been described in canine histiocytic diseases.
LCH is clinically divided into three groups: unifocal, multifocal unisystem, and multifocal multisystem.[37]
Unifocal
Unifocal LCH, also called eosinophilic granuloma (an older term which is now known to be a misnomer), is a disease characterized by an expanding proliferation of Langerhans cells in one organ, where they cause damage called lesions. It typically has no extraskeletal involvement, but rarely a lesion can be found in the skin, lungs, or stomach. It can appear as a single lesion in an organ, up to a large quantity of lesions in one organ. When multiple lesions are scattered throughout an organ, it can be called a multifocal unisystem variety. When found in the lungs, it should be distinguished from Pulmonary Langerhans cell hystiocytosis—a special category of disease most commonly seen in adult smokers.[38] When found in the skin it is called cutaneous single system Langerhans cell LCH. This version can heal without therapy in some rare cases.[39] This primary bone involvement helps to differentiate eosinophilic granuloma from other forms of Langerhans Cell Histiocytosis (Letterer-Siwe or Hand-Schüller-Christian variant).[40]
Multifocal unisystem
Seen mostly in children, multifocal unisystem LCH is characterized by fever, bone lesions and diffuse eruptions, usually on the scalp and in the ear canals. 50% of cases involve the pituitary stalk, often leading to diabetes insipidus. The triad of diabetes insipidus, exophthalmos, and lytic bone lesions is known as the chronic multifocal Langerhans cell histiocytosis. Peak onset is 2–10 years of age.[citation needed]
Multifocal multisystem
Multifocal multisystem LCH, also called Letterer-Siwe disease, is an often rapidly progressing disease in which Langerhans Cell cells proliferate in many tissues. It is mostly seen in children under age 2, and the prognosis is poor: even with aggressive chemotherapy, the five-year survival is only 50%.[41]
Lungs
Pulmonary Langerhans cell histiocytosis (PLCH) is a unique form of LCH in that it occurs almost exclusively in cigarette smokers. It is now considered a form of smoking-related interstitial lung disease. PLCH develops when an abundance of monoclonal CD1a-positive Langerhans (immature histiocytes) proliferate the bronchioles and alveolar interstitium, and this flood of histiocytes recruits granulocytes like eosinophils and neutrophils and agranulocytes like lymphocytes further destroying bronchioles and the interstitial alveolar space that can cause damage to the lungs.[42] It is hypothesized that bronchiolar destruction in PLCH is first attributed to the special state of Langerhans cells that induce cytotoxic T-cell responses, and this is further supported by research that has shown an abundance of T-cells in early PLCH lesions that are CD4+ and present early activation markers. [43]Some affected people recover completely after they stop smoking, but others develop long-term complications such as pulmonary fibrosis and pulmonary hypertension.[44] PLCH patients, families, and caregivers are encouraged to join the NIH Rare Lung Diseases Consortium Contact Registry Archived 2019-03-27 at the Wayback Machine. This is a privacy protected site that provides up-to-date information for individuals interested in the latest scientific news, trials, and treatments related to rare lung diseases.[citation needed]
Treatment
Guidelines for management of patients up to 18 years with Langerhans cell histiocytosis have been suggested.[45][46][47][48] Treatment is guided by extent of disease. Solitary bone lesion may be amenable through excision or limited radiation, dosage of 5-10 Gy for children, 24-30 Gy for adults. However systemic diseases often require chemotherapy. Use of systemic steroid is common, singly or adjunct to chemotherapy. Local steroid cream is applied to skin lesions. Endocrine deficiency often require lifelong supplement e.g. desmopressin for diabetes insipidus which can be applied as nasal drop. Chemotherapeutic agents such as alkylating agents, antimetabolites, vinca alkaloids either singly or in combination can lead to complete remission in diffuse disease.[citation needed]
Prognosis
Excellent for single-focus disease. With multi-focal disease 60% have a chronic course, 30% achieve remission and mortality is up to 10%.[49]
Epidemiology
LCH usually affects children between 1 and 15 years old, with a peak incidence between 5 and 10 years of age. Among children under the age of 10, yearly incidence is thought to be 1 in 200,000;[50] and in adults even rarer, in about 1 in 560,000.[51] It has been reported in elderly but is vanishingly rare.[52] It is most prevalent in Caucasians, and affects males twice as often as females.[53] In other populations the rates in males is slightly more than in females.[54]
History
MRI and CT scan findings in a mummy have revealed evidence of the disease dating back to 900-790. B.C.[7]
The subtype chronic multifocal Langerhans cell histiocytosis was previously known as Hand–Schüller–Christian disease, and was named for the American pediatrician Alfred Hand Jr.,[9] the Austrian neuroradiologist Arthur Schüller,[9][55] and the American pathologist Henry Asbury Christian,[9][56] who described it in 1893,[9][57] 1915[9][58] and 1919, respectively.[9][59] Shortly afterwards, Letterer in 1924 and Siwe in 1933 described a fatal condition in children who presented with large livers and spleens, large lymph nodes and bone damage. In 1940, Louis Litchtenstein and Henry L. Jaffe described a self-limiting disease characterised by "isolated bone lesions".[60] A common feature of all these conditions was revealed to be the histological findings of large numbers of histiocytes in the tissue biopsies, leading Litchtenstein to propose that the three described conditions were part of a spectrum of a disease he named "Histiocytosis X", where "X" denoted the unknown cause at the time.[60] In 1973, Christian Nezelof recognised the abnormal cell as a 'Langerhans-like' cell, however it took another ten years for the term 'Langerhans cell histiocytosis' to be internationally recognised. In 1987, the Histiocyte Society published their classification of the histiocyte disorders together with criteria for diagnosis and clinical assessment of Langerhans cell histiocytosis.[61]
Langerhans cell histiocytosis is occasionally misspelled as "Langerhan" or "Langerhan's" cell histiocytosis, even in authoritative textbooks. The name, however, originates back to its discoverer, Paul Langerhans.[62]
Society and culture
In the 10th episode of season 3 of House entitled "Merry Little Christmas", the primary patient is a girl with dwarfism who has a variety of symptoms, who is ultimately diagnosed with Langerhans cell histiocytosis.[63]Also in the 5th episode, season 1 of "The Good Doctor", Dr. Murphy tries to diagnose Langerhans cell histiocytosis in a boy with a previously diagnosed osteosarcoma.[citation needed]In an episode of Mystery Diagnosis, "The Woman Who Saw Pink", Brooke Rohrer has experiencing symptoms of abdominal pain, was diagnosed with Langerhans cell histiocytosis.[citation needed]
References
- ↑ 1.00 1.01 1.02 1.03 1.04 1.05 1.06 1.07 1.08 1.09 1.10 1.11 1.12 1.13 1.14 1.15 1.16 1.17 1.18 1.19 1.20 1.21 "Langerhans Cell Histiocytosis". NORD (National Organization for Rare Disorders). Archived from the original on 10 June 2021. Retrieved 5 December 2020.
- ↑ 2.0 2.1 2.2 2.3 2.4 2.5 2.6 2.7 2.8 "Langerhans cell histiocytosis". rarediseases.info.nih.gov. National Institutes of Health. Archived from the original on 10 January 2021. Retrieved 6 December 2020.
- ↑ 3.0 3.1 3.2 "Orphanet: Langerhans cell histiocytosis". www.orpha.net. Archived from the original on 28 August 2021. Retrieved 5 December 2020.
- ↑ 4.0 4.1 4.2 4.3 4.4 Payne, Jacqueline. "Langerhans' Cell Histiocytosis". patient.info. Archived from the original on 12 February 2012. Retrieved 6 December 2020. Last updated 28 Feb 2017.
- ↑ Tebbi, Cameron K. (16 September 2020). Kanwar, Vikramjit S (ed.). "What is the Histiocyte Society classification of histiocytosis syndromes?". Medscape. Archived from the original on 20 January 2021. Retrieved 7 December 2020.
- ↑ 6.0 6.1 6.2 "Langerhans cell histiocytosis: MedlinePlus Genetics". medlineplus.gov. MedlinePlus. Archived from the original on 10 January 2021. Retrieved 6 December 2020.
- ↑ 7.0 7.1 Cavka, Mislav; Petaros, Anja; Ivanac, Gordana; Aganović, Lejla; Janković, Ivor; Reiter, Gert; Speier, Peter; Nielles-Vallespin, Sonja; Brkljacić, Boris (March 2012). "A probable case of Hand-Schueller-Christian's disease in an Egyptian mummy revealed by CT and MR investigation of a dry mummy". Collegium Antropologicum. 36 (1): 281–286. ISSN 0350-6134. PMID 22816232.
- ↑ Broadbent, V.; Egeler, R. M.; Nesbit, M. E. (September 1994). "Langerhans cell histiocytosis--clinical and epidemiological aspects". The British Journal of Cancer. Supplement. 23: S11–S16. ISSN 0306-9443. PMC 2149699. PMID 8075001. Archived from the original on 2021-08-28. Retrieved 2020-12-06.
- ↑ 9.0 9.1 9.2 9.3 9.4 9.5 9.6 Pluot, Etienne; Davies, Mark; James, Steven L. J. (2009). "Who was who in bone tumours". In Davies, A. Mark; Sundaram, Murali; James, Steven J. (eds.). Imaging of Bone Tumors and Tumor-Like Lesions: Techniques and Applications. Springer. p. 681. ISBN 978-3-540-77982-7. Archived from the original on 2021-08-28. Retrieved 2020-12-06.
- ↑ Kaltsas, GA; Powles, TB; Evanson, J; Plowman, PN; Drinkwater, JE; Jenkins, PJ; Monson, JP; Besser, GM; Grossman, AB (April 2000). "Hypothalamo-pituitary abnormalities in adult patients with langerhans cell histiocytosis: clinical, endocrinological, and radiological features and response to treatment". The Journal of Clinical Endocrinology and Metabolism. 85 (4): 1370–6. doi:10.1210/jcem.85.4.6501. PMID 10770168.
- ↑ Grois, N; Pötschger, U; Prosch, H; Minkov, M; Arico, M; Braier, J; Henter, JI; Janka-Schaub, G; Ladisch, S; Ritter, J; Steiner, M; Unger, E; Gadner, H; DALHX- and LCH I and II Study, Committee (February 2006). "Risk factors for diabetes insipidus in langerhans cell histiocytosis". Pediatric Blood & Cancer. 46 (2): 228–33. doi:10.1002/pbc.20425. PMID 16047354.
- ↑ Broadbent, V; Dunger, DB; Yeomans, E; Kendall, B (1993). "Anterior pituitary function and computed tomography/magnetic resonance imaging in patients with Langerhans cell histiocytosis and diabetes insipidus". Medical and Pediatric Oncology. 21 (9): 649–54. doi:10.1002/mpo.2950210908. PMID 8412998.
- ↑ Cisternino A, Asa'ad F, Fusco N, Ferrero S, Rasperini G (Oct 2015). "Role of multidisciplinary approach in a case of Langerhans cell histiocytosis with initial periodontal manifestations". Int J Clin Exp Pathol. 8 (10): 13539–45. PMC 4680515. PMID 26722570.
- ↑ Sholl, LM; Hornick, JL; Pinkus, JL; Pinkus, GS; Padera, RF (June 2007). "Immunohistochemical analysis of langerin in langerhans cell histiocytosis and pulmonary inflammatory and infectious diseases". The American Journal of Surgical Pathology. 31 (6): 947–52. doi:10.1097/01.pas.0000249443.82971.bb. PMID 17527085. S2CID 19702745.
- ↑ Stanway, Amy (2005). "Langerhans cell histiocytosis | DermNet NZ". dermnetnz.org. Archived from the original on 14 August 2021. Retrieved 4 December 2020.
- ↑ Kapur, Payal; Erickson, Christof; Rakheja, Dinesh; Carder, K. Robin; Hoang, Mai P. (2007). "Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease): Ten-year experience at Dallas Children's Medical Center". Journal of the American Academy of Dermatology. 56 (2): 290–4. doi:10.1016/j.jaad.2006.09.001. PMID 17224372.
- ↑ "Langerhans Cell Histiocytosis Treatment (PDQ®)–Patient Version - National Cancer Institute". www.cancer.gov. 16 July 2010. Archived from the original on 29 October 2021. Retrieved 5 December 2020.
- ↑ "Langerhans' cell histiocytosis". www.gosh.nhs.uk. Archived from the original on 24 May 2021. Retrieved 5 December 2020.
- ↑ Broadbent, V.; Davies, E.G.; Heaf, D.; Pincott, J.R.; Pritchard, J.; Levinsky, R.J.; Atherton, D.J.; Tucker, S. (1984). "Spontaneous remission of multi-system histiocytosis X". Lancet. 1 (8371): 253–4. doi:10.1016/S0140-6736(84)90127-2. PMID 6142997. S2CID 46217705.
- ↑ Filippi, Paola; De Badulli, Carla; Cuccia, Mariaclara; Silvestri, Annalisa; De Dametto, Ennia; Pasi, Annamaria; Garaventa, Alberto; Prever, Adalberto Brach; del Todesco, Alessandra; Trizzino, Antonino; Danesino, Cesare; Martinetti, Miryam; Arico, Maurizio (2006). "Specific polymorphisms of cytokine genes are associated with different risks to develop single-system or multi-system childhood Langerhans cell histiocytosis". British Journal of Haematology. 132 (6): 784–7. doi:10.1111/j.1365-2141.2005.05922.x. PMID 16487180.
- ↑ Steen, A.E.; Steen, K.H.; Bauer, R.; Bieber, T. (1 July 2001). "Successful treatment of cutaneous Langerhans cell histiocytosis with low-dose methotrexate". British Journal of Dermatology. 145 (1): 137–140. doi:10.1046/j.1365-2133.2001.04298.x. PMID 11453923.
- ↑ Allen CE, Flores R, Rauch R, Dauser R, Murray JC, Puccetti D, Hsu DA, Sondel P, Hetherington M, Goldman S, McClain KL (2010). "Neurodegenerative central nervous system Langerhans cell histiocytosis and coincident hydrocephalus treated with vincristine/cytosine arabinoside". Pediatr Blood Cancer. 54 (3): 416–23. doi:10.1002/pbc.22326. PMC 3444163. PMID 19908293.
- ↑ Minkov, M.; Grois, N.; Broadbent, V.; Ceci, A.; Jakobson, A.; Ladisch, S. (1 November 1999). "Cyclosporine A therapy for multisystem Langerhans cell histiocytosis". Medical and Pediatric Oncology. 33 (5): 482–485. doi:10.1002/(SICI)1096-911X(199911)33:5<482::AID-MPO8>3.0.CO;2-Y. PMID 10531573.
- ↑ Willman, Cheryl L.; Busque, Lambert; Griffith, Barbara B.; Favara, Blaise E.; McClain, Kenneth L.; Duncan, Marilyn H.; Gilliland, D. Gary (21 July 1994). "Langerhans'-Cell Histiocytosis (Histiocytosis X) -- A Clonal Proliferative Disease". New England Journal of Medicine. 331 (3): 154–160. doi:10.1056/NEJM199407213310303. PMID 8008029.
- ↑ Badalian-Very G, Vergilio JA, Degar BA, MacConaill LE, Brandner B, Calicchio ML, Kuo FC, Ligon AH, Stevenson KE, Kehoe SM, Garraway LA, Hahn WC, Meyerson M, Fleming MD, Rollins BJ (2010). "Recurrent BRAF mutations in Langerhans cell histiocytosis". Blood. 116 (11): 1919–23. doi:10.1182/blood-2010-04-279083. PMC 3173987. PMID 20519626.
- ↑ Satoh T, Smith A, Sarde A, Lu HC, Mian S, Mian S, Trouillet C, Mufti G, Emile JF, Fraternali F, Donadieu J, Geissmann F (2012). "B-RAF mutant alleles associated with Langerhans cell histiocytosis, a granulomatous pediatric disease". PLOS ONE. 7 (4): e33891. Bibcode:2012PLoSO...733891S. doi:10.1371/journal.pone.0033891. PMC 3323620. PMID 22506009.
- ↑ Peters, Tricia L.; Tsz-Kwong Chris Man; Price, Jeremy; George, Renelle; Phaik Har Lim; Kenneth Matthew Heym; Merad, Miriam; McClain, Kenneth L.; Allen, Carl E. (10 December 2011). "1372 Frequent BRAF V600E Mutations Are Identified in CD207+ Cells in LCH Lesions, but BRAF Status does not Correlate with Clinical Presentation of Patients or Transcriptional Profiles of CD207+ Cells". Archived from the original on 26 July 2014. Retrieved 28 June 2012.
Oral and Poster Abstracts presented at 53rd ASH Annual Meeting and Exposition
{{cite journal}}: Cite journal requires|journal=(help) - ↑ Monsereenusorn, Chalinee; Rodriguez-Galindo, Carlos (October 2015). "Clinical Characteristics and Treatment of Langerhans Cell Histiocytosis". Hematology/Oncology Clinics of North America. 29 (5): 853–873. doi:10.1016/j.hoc.2015.06.005. PMID 26461147.
- ↑ 29.0 29.1 Wilson, AJ; Maddox, PH; Jenkins, D (January 1991). "CD1a and S100 antigen expression in skin Langerhans cells in patients with breast cancer". The Journal of Pathology. 163 (1): 25–30. doi:10.1002/path.1711630106. PMID 2002421.
- ↑ Coppola, D; Fu, L; Nicosia, SV; Kounelis, S; Jones, M (May 1998). "Prognostic significance of p53, bcl-2, vimentin, and S100 protein-positive Langerhans cells in endometrial carcinoma". Human Pathology. 29 (5): 455–62. doi:10.1016/s0046-8177(98)90060-0. PMID 9596268.
- ↑ McLelland, J; Chu, AC (October 1988). "Comparison of peanut agglutinin and S100 stains in the paraffin tissue diagnosis of Langerhans cell histiocytosis". The British Journal of Dermatology. 119 (4): 513–9. doi:10.1111/j.1365-2133.1988.tb03255.x. PMID 2461216.
- ↑ Ye, F; Huang, SW; Dong, HJ (November 1990). "Histiocytosis X. S-100 protein, peanut agglutinin, and transmission electron microscopy study". American Journal of Clinical Pathology. 94 (5): 627–31. doi:10.1093/ajcp/94.5.627. PMID 2239828.
- ↑ Valladeau J, Ravel O, Dezutter-Dambuyant C, Moore K, Kleijmeer M, Liu Y, Duvert-Frances V, Vincent C, Schmitt D, Davoust J, Caux C, Lebecque S, Saeland S (2000). "Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules". Immunity. 12 (1): 71–81. doi:10.1016/S1074-7613(00)80160-0. PMID 10661407.
- ↑ Lau, SK; Chu, PG; Weiss, LM (April 2008). "Immunohistochemical expression of Langerin in Langerhans cell histiocytosis and non-Langerhans cell histiocytic disorders". The American Journal of Surgical Pathology. 32 (4): 615–9. doi:10.1097/PAS.0b013e31815b212b. PMID 18277880. S2CID 40092500.
- ↑ "Orphanet: OBSOLETE: Hand Schüller Christian disease". www.orpha.net. Archived from the original on 15 November 2017. Retrieved 5 December 2020.
- ↑ Makras P, Papadogias D, Kontogeorgos G, Piaditis G, Kaltsas G (2005). "Spontaneous gonadotrophin deficiency recovery in an adult patient with Langerhans cell Histiocytosis (LCH)". Pituitary. 8 (2): 169–74. doi:10.1007/s11102-005-4537-z. PMID 16379033. S2CID 7878051.
- ↑ Cotran, Ramzi S.; Kumar, Vinay; Abbas, Abul K.; Fausto, Nelson; Robbins, Stanley L. (2005). Robbins and Cotran pathologic basis of disease. St. Louis, Mo: Elsevier Saunders. pp. 701–. ISBN 978-0-8089-2302-2.
- ↑ Kumar, Vinay; Abbas, Abul; Aster, Jon (2015). Robbins and Cotran Pathologic Basis of Disease (9th ed.). Philadelphia, PA: Elsevier. p. 622. ISBN 978-1-4557-2613-4.
- ↑ Afsar, Fatma Sule; Ergin, Malik; Ozek, Gulcihan; Vergin, Canan; Karakuzu, Ali; Seremet, Sila (2017). "Histiocitose de Células de Langerhans Autolimitada e de Início Tardio: Relato de Uma Entidade Raríssima". Revista Paulista de Pediatria. 35 (1): 115–119. doi:10.1590/1984-0462/;2017;35;1;00015. ISSN 0103-0582. PMC 5417814. PMID 28977321.
- ↑ Ladisch, Stephan (2011). "Histiocytosis Syndromes of Childhood". In Kliegman, Robert M.; Stanton, Bonita F.; St. Geme, Joseph; Schor, Nina; Behrman, Richard E. (eds.). Nelson Textbook of Pediatrics (19th ed.). Saunders. pp. 1773–7. ISBN 978-1-4377-0755-7.
- ↑ Langerhans Cell Histiocytosis at eMedicine
- ↑ Juvet, Stephan. "Rare lung diseases III: Pulmonary Langerhans' cell histiocytosis". PubMed Central. Canadian Respiratory Journal. Archived from the original on 2021-04-20. Retrieved 2020-11-28.
- ↑ Tazi, A. "Adult pulmonary Langerhans' cell histiocytosis". European Respiratory Journal. European Respiratory Journal. Archived from the original on 2020-10-31. Retrieved 2020-11-30.
- ↑ Juvet, Stephen C; Hwang, David; Downey, Gregory P (2010). "Rare lung diseases III: pulmonary Langerhans' cell histiocytosis". Canadian Respiratory Journal. 17 (3): e55–62. doi:10.1155/2010/216240. PMC 2900147. PMID 20617216.
- ↑ Haupt, Riccardo; Minkov, Milen; Astigarraga, Itziar; Schäfer, Eva; Nanduri, Vasanta; Jubran, Rima; Egeler, R. Maarten; Janka, Gritta; Micic, Dragan; Rodriguez-Galindo, Carlos; Van Gool, Stefaan; Visser, Johannes; Weitzman, Sheila; Donadieu, Jean (February 2013). "Langerhans cell histiocytosis (LCH): Guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years". Pediatric Blood & Cancer. 60 (2): 175–184. doi:10.1002/pbc.24367. PMC 4557042. PMID 23109216.
- ↑ Girschikofsky, Michael; Arico, Maurizio; Castillo, Diego; Chu, Anthony; Doberauer, Claus; Fichter, Joachim; Haroche, Julien; Kaltsas, Gregory A; Makras, Polyzois; Marzano, Angelo V; de Menthon, Mathilde; Micke, Oliver; Passoni, Emanuela; Seegenschmiedt, Heinrich M; Tazi, Abdellatif; McClain, Kenneth L (2013). "Management of adult patients with Langerhans cell histiocytosis: recommendations from an expert panel on behalf of Euro-Histio-Net". Orphanet Journal of Rare Diseases. 8 (1): 72. doi:10.1186/1750-1172-8-72. PMC 3667012. PMID 23672541.
- ↑ "Langerhans cell histiocytosis - Histiocyte Society Evaluation and Treatment Guidelines". April 2009. Archived from the original on 2018-01-21. Retrieved 2015-04-28.
{{cite journal}}: Cite journal requires|journal=(help) - ↑ "Langerhans Cell Histiocytosis Treatment (PDQ®)". Archived from the original on 2021-08-28. Retrieved 2017-12-02.
{{cite journal}}: Cite journal requires|journal=(help) and 1 Archived 2015-04-26 at the Wayback Machine - ↑ Komp D, El Mahdi A, Starling K, Easley J, Vietti T, Berry D, George S (1980). "Quality of survival in Histiocytosis X: a Southwest Oncology Group study". Med. Pediatr. Oncol. 8 (1): 35–40. doi:10.1002/mpo.2950080106. PMID 6969347.
- ↑ "MedlinePlus Medical Encyclopedia: Histiocytosis". Archived from the original on 2016-07-04. Retrieved 2007-05-10.
- ↑ "Histiocytosis Association of Canada". Archived from the original on 2007-05-14. Retrieved 2007-05-16.
- ↑ Gerlach, Beatrice; Stein, Annette; Fischer, Rainer; Wozel, Gottfried; Dittert, Dag-Daniel; Richter, Gerhard (1998). "Langerhanszell-Histiozytose im Alter" [Langerhans cell histiocytosis in the elderly]. Der Hautarzt (in Deutsch). 49 (1): 23–30. doi:10.1007/s001050050696. PMID 9522189. S2CID 20706403.
- ↑ Sellari-Franceschini, S.; Forli, F.; Pierini, S.; Favre, C.; Berrettini, S.; Macchia, P.A. (1999). "Langerhans' cells histiocytosis: a case report". International Journal of Pediatric Otorhinolaryngology. 48 (1): 83–7. doi:10.1016/S0165-5876(99)00013-0. PMID 10365975.
- ↑ Aricò, M.; Girschikofsky, M.; Généreau, T.; Klersy, C.; McClain, K.; Grois, N.; Emile, J.-F.; Lukina, E.; De Juli, E.; Danesino, C. (2003). "Langerhans cell histiocytosis in adults: Report from the International Registry of the Histiocyte Society". European Journal of Cancer. 39 (16): 2341–8. doi:10.1016/S0959-8049(03)00672-5. PMID 14556926.
- ↑ Schindler, E. (August 1997). "Arthur Schüller: pioneer of neuroradiology". AJNR. American journal of neuroradiology. 18 (7): 1297–1302. ISSN 0195-6108. PMID 9282858. Archived from the original on 2021-08-28. Retrieved 2020-12-06.
- ↑ "Obituary; Henry Asbury Christian". New England Journal of Medicine. 245 (23): 912–913. 6 December 1951. doi:10.1056/NEJM195112062452312. ISSN 0028-4793. PMID 14882449. Archived from the original on 28 August 2021. Retrieved 6 December 2020.
- ↑ A. Hand. Polyuria and tuberculosis. Proceedings of the Pathological Society of Philadelphia, 1893, 16: 282-284. Archives of Pediatrics, New York, 1893: 10: 673-675.
- ↑ Schüller, Arthur (1915). Über eigenartige Schädeldefekte im Jugendalter, Fortschr. a. d. Geb. d. Röentgenstrahlen 23: 12–18.
- ↑ H. Christian. Defects in membranous bones, exophthalmos, and diabetes insipidus; an unusual syndrome of dyspituitarism. In: Contributions to medical and biological research, dedicated to Sir William Osler. New York, P. B. Hoeber, 1919, 1: 390-401. Medical Clinics of North America, Philadelphia, PA., 1920; 3: 849-871.
- ↑ 60.0 60.1 C.A.C. Pickering; L. Doyle; K.B. Carroll (1981). "4. Honeycomb lung". Interstitial Lung Disease. MTP Press. p. 83-85. ISBN 978-94-009-8086-0. Archived from the original on 2021-08-28. Retrieved 2020-12-06.
- ↑ Broadbent, V.; Egeler, R. M.; Nesbit, M. E. (September 1994). "Langerhans cell histiocytosis--clinical and epidemiological aspects". The British Journal of Cancer. Supplement. 23: S11–S16. ISSN 0306-9443. PMC 2149699. PMID 8075001. Archived from the original on 2021-08-28. Retrieved 2020-12-06.
- ↑ Jolles, S. (April 2002). "Paul Langerhans". Journal of Clinical Pathology. 55 (4): 243. doi:10.1136/jcp.55.4.243. PMC 1769627. PMID 11919207.
- ↑ House (season 3) In CSI, season 15 episode 4, a student has LCH.
External links
| Classification | |
|---|---|
| External resources |
- Pages with script errors
- CS1 errors: missing periodical
- Webarchive template wayback links
- CS1 Deutsch-language sources (de)
- All articles with unsourced statements
- Articles with unsourced statements from September 2020
- Articles with invalid date parameter in template
- Articles with unsourced statements from July 2020
- Articles with unsourced statements from April 2018
- Histiocytosis
- Monocyte- and macrophage-related cutaneous conditions
- Rare diseases
- RTT