Hirschsprung's disease
| Hirschsprung's disease | |
|---|---|
| Other names: Hirschsprung-associated enterocolitis,[1] aganglionic megacolon, congenital megacolon, congenital intestinal aganglionosis[2] | |
 | |
| Histopathology of Hirschsprung disease showing abnormal acetylcholine esterase (AchE)-positive nerve fibers (brown) in the mucosa | |
| Specialty | Medical genetics |
| Symptoms | Constipation, vomiting, abdominal pain, diarrhea, slow growth[2] |
| Complications | Enterocolitis, megacolon, bowel obstruction, intestinal perforation[2][3] |
| Usual onset | First 2 months of life[2] |
| Types | Short-segment, long-segment[2] |
| Causes | Genetic[2] |
| Risk factors | Family history[2] |
| Diagnostic method | Based on symptoms, biopsy[1] |
| Differential diagnosis | Chronic intestinal pseudo-obstruction, meconium ileus[3] |
| Treatment | Surgery[3] |
| Frequency | 1 in 5,000 newborns[2] |
Hirschsprung's disease (HD or HSCR) is a birth defect in which nerves are missing from parts of the intestine.[2][1] The most prominent symptom is constipation.[2] Other symptoms may include vomiting, abdominal pain, diarrhea and slow growth.[2] Symptoms usually become apparent in the first two months of life.[2] Complications may include enterocolitis, megacolon, bowel obstruction and intestinal perforation.[2][3]
The disorder may occur by itself or in association with other genetic disorders such as Down syndrome or Waardenburg syndrome.[2][3] About half of isolated cases are linked to a specific genetic mutation, and about 20% occur within families.[2] Some of these occur in an autosomal dominant manner.[2] The cause of the remaining cases is unclear.[2] If otherwise normal parents have one child with the condition, the next child has a 4% risk of being affected.[3] The condition is divided into two main types, short-segment and long-segment, depending on how much of the bowel is affected.[2] Rarely, the small bowel may be affected, as well.[3] Diagnosis is based on symptoms and confirmed by biopsy.[1]
Treatment is generally by surgery to remove the affected section of bowel.[3] The surgical procedure most often carried out is known as a "pull through".[1] Occasionally, an intestinal transplantation may be recommended.[3] Hirschsprung's disease occurs in about one in 5,000 of newborns.[2] Males are more often affected than females.[2] The condition is believed to have first been described in 1691 by Dutch anatomist Frederik Ruysch[4] and is named after Danish physician Harald Hirschsprung following his description in 1888.[5][6]
Signs and symptoms
Typically, Hirschsprung disease is diagnosed shortly after birth, although it may develop well into adulthood, because of the presence of megacolon, or because the baby fails to pass the first stool (meconium)[7] within 48 hours of delivery. Normally, 90% of babies pass their first meconium within 24 hours, and 99% within 48 hours.[8] Other symptoms include green or brown vomit, explosive stools after a doctor inserts a finger into the rectum, swelling of the abdomen, excessive gas, and bloody diarrhea.
Some cases are diagnosed later, into childhood, but usually before age 10.[7] The child may experience fecal retention, constipation, or abdominal distention.[7]
Cause
The disorder may occur by itself or in association with other genetic disorders such as Down syndrome.[3] About half of isolated cases are linked to a specific genetic mutation and about 20% occur within families.[2] Some of these occur in an autosomal dominant manner.[2] The cause of the remaining cases is unclear.[2] If otherwise normal parents have one child with the condition, the next child has a 4% risk of being affected.[3]
Genetics
| Type | OMIM | Gene | Locus |
|---|---|---|---|
| HSCR1 | 142623 | RET | 10q11.2 |
| HSCR2 | 600155 | EDNRB | 13q22 |
| HSCR3 | 600837 | GDNF | 5p13.1-p12 |
| HSCR4 | 131242 | EDN3 | 20q13.2-q13.3 |
| HSCR5 | 600156 | ? | 21q22 |
| HSCR6 | 606874 | ? | 3p21 |
| HSCR7 | 606875 | ? | 19q12 |
| HSCR8 | 608462 | ? | 16q23 |
| HSCR9 | 611644 | ? | 4q31-32 |
| — | 602229 | SOX10 | 22q13 |
| — | 600423 | ECE1 | 1p36.1 |
| — | 602018 | NRTN | 19p13.3 |
| — | 602595 | GEMIN2 (Gem-associated protein 2) | 14q13-q21 |
| — | 191315 | NTRK1 | 1q23.1 |
| — | 605802 | ZEB2 | 2q22.3 |
Several genes and specific regions on chromosomes (loci) have been shown or suggested to be associated with Hirschsprung's disease:
The RET proto-oncogene accounts for the highest proportion of both familial and sporadic cases, with a wide range of mutations scattered along its entire coding region.[9] A proto-oncogene can cause cancer if it is mutated or overexpressed.
RET proto-oncogene
RET is a gene that codes for proteins that assist cells of the neural crest in their movement through the digestive tract during the development of the embryo. Those neural crest cells eventually form bundles of nerve cells called ganglions. EDNRB codes for proteins that connect these nerve cells to the digestive tract. Thus, mutations in these two genes could directly lead to the absence of certain nerve fibers in the colon. Research suggests that several genes are associated with Hirschsprung’s disease.[10] Also, new research suggests that mutations in genomic sequences involved in regulating EDNRB have a bigger impact on Hirschsprung’s disease than previously thought.[citation needed]
RET can mutate in many ways and is associated with Down syndrome. Since Down syndrome is comorbid in 2% of Hirschsprung’s cases, a likelihood exists that RET is involved heavily in both Hirschsprung's disease and Down syndrome. RET is also associated with medullary thyroid cancer and neuroblastoma, which is a type of cancer common in children. Both of these disorders are more common in Hirschsprung’s patients than in the general population. One function that RET controls is the travel of the neural crest cells through the intestines in the developing fetus. The earlier the RET mutation occurs in Hirschsprung’s disease, the more severe the disorder becomes.
Other genes
Common and rare DNA variations in the neuregulin 1 (NRG1) and NRG3 (NRG3) were first shown to be associated with the disease in Chinese patients through a Genome Wide Association Study by the Hong Kong team in 2009 [11] and 2012, respectively[12] Subsequent studies in both Asian and Caucasian patients confirmed the initial findings by the University of Hong Kong. Both rare and common variants in these two genes have been identified in additional Chinese,[13] Thai, Korean, Indonesian, and Spanish patients. These two genes are known to play a role in the formation of the enteric nervous system; thus, they are likely to be involved in the pathology of Hirschsprung's disease, at least in some cases.[citation needed]
Another gene associated with this condition is NADPH oxidase, EF-hand calcium binding domain 5 (NOX5).[14] This gene is located on the long arm of chromosome 15 (15q23).
Associated syndromes
Hirschsprung's disease can also present as part of multi system disorders, such as:[15]
- Bardet–Biedl syndrome
- Cartilage–hair hypoplasia[16]
- Congenital central hypoventilation syndrome[17]
- MEN2[18]
- Mowat–Wilson syndrome[19]
- Smith–Lemli–Opitz syndrome[20]
- Trisomy 21 (Down syndrome) [21]
- Some forms of Waardenburg syndrome
Pathophysiology

During normal prenatal development, cells from the neural crest migrate into the large intestine (colon) to form the networks of nerves called the myenteric plexus (Auerbach plexus) (between the smooth muscle layers of the gastrointestinal tract wall) and the submucosal plexus (Meissner plexus) (within the submucosa of the gastrointestinal tract wall). In Hirschsprung disease, the migration is not complete and part of the colon lacks these nerve bodies that regulate the activity of the colon. The affected segment of the colon cannot relax and pass stool through the colon, creating an obstruction.[23]
The most accepted theory of the cause of Hirschsprung is a defect in the craniocaudal migration of neuroblasts originating from the neural crest that occurs during the first 12 weeks of gestation. Defects in the differentiation of neuroblasts into ganglion cells and accelerated ganglion cell destruction within the intestine may also contribute to the disorder.[24]
This lack of ganglion cells in the myenteric and submucosal plexus is well documented in Hirschsprung's disease.[7] With Hirschsprung's disease, the segment lacking neurons (aganglionic) becomes constricted, causing the normal, proximal section of bowel to become distended with feces. This narrowing of the distal colon and the failure of relaxation in the aganglionic segment are thought to be caused by the lack of neurons containing nitric oxide synthase.[7]
The most cited feature is absence of ganglion cells: notably in males, 75% have none in the end of the colon (rectosigmoid) and 8% lack ganglion cells in the entire colon. The enlarged section of the bowel is found proximally, while the narrowed, aganglionic section is found distally, closer to the end of the bowel. The absence of ganglion cells results in a persistent overstimulation of nerves in the affected region, resulting in contraction.
The equivalent disease in horses is lethal white syndrome.[25]
Diagnosis
Definitive diagnosis is made by suction biopsy of the distally narrowed segment.[26] A histologic examination of the tissue would show a lack of ganglionic nerve cells. Diagnostic techniques involve anorectal manometry,[27] barium enema, and rectal biopsy. The suction rectal biopsy is considered the current international gold standard in the diagnosis of Hirschsprung's disease.[28]
Radiologic findings may also assist with diagnosis.[29] Cineanography (fluoroscopy of contrast medium passing anorectal region) assists in determining the level of the affected intestines.
-
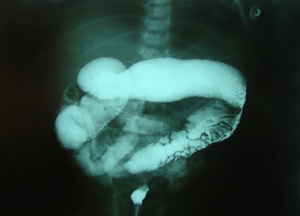
Barium enema in neonate with Hirschsprung disease
-

A: Plain abdominal radiograph showing a PARTZ at rectosigmoid, arrow. B: Plain abdominal radiograph showing a PARTZ at midsigmoid, arrow. C: Plain abdominal radiograph showing a PARTZ at descending colon, arrow. D: Contrast enema showing a CETZ at rectosigmoid, arrow. E: Contrast enema showing a CETZ at midsigmoid, arrow. F: Contrast enema showing a CETZ at descending colon, arrow.


Treatment
Treatment of Hirschsprung's disease consists of surgical removal (resection) of the abnormal section of the colon, followed by reanastomosis.
Colostomy
The first stage of treatment used to be a reversible colostomy. In this approach, the healthy end of the large intestine is cut and attached to an opening created on the front of the abdomen. The contents of the bowel are discharged through the hole in the abdomen and into a bag. Later, when the patient's weight, age, and condition are right, the "new" functional end of the bowel is connected with the anus. The first surgical treatment involving surgical resection followed by reanastomosis without a colostomy occurred as early as 1933 by Doctor Baird in Birmingham on a one-year-old boy.
Other procedures
The Swedish-American surgeon, Orvar Swenson (1909–2012), who discovered the cause of Hirschsprung’s, first performed its surgical treatment, the pull-through surgery, in 1948.[30] The pull-through procedure repairs the colon by connecting the functioning portion of the bowel to the anus. The pull-through procedure is the typical method for treating Hirschsprung’s in younger patients. Swenson devised the original procedure, and the pull-through surgery has been modified many times.[31]'
Currently, several different surgical approaches are used, which include the Swenson, Soave, Duhamel, and Boley procedures.[31] The Swenson procedure leaves a small portion of the diseased bowel. The Soave procedure, named after the Italian pediatric surgeon, Franco Soave (1917–1984), leaves the outer wall of the colon unaltered. The Boley procedure, pioneered by the American surgeon, Scott Boley (b. 1941), is a small modification of the Soave procedure, so the term "Soave-Boley" procedure is sometimes used.[32][33] The Duhamel procedure, named for the French pediatric surgeon, Bernard Duhamel (1917–1996), uses a surgical stapler to connect the good and bad bowel.
For the 15% of children who do not obtain full bowel control, other treatments are available. Constipation may be remedied by laxatives or a high-fiber diet. In those patients, serious dehydration can play a major factor in their lifestyles. A lack of bowel control may be addressed by an ileostomy – similar to a colostomy, but uses the end of the small intestine rather than the colon. The Malone antegrade colonic enema (ACE) is also an option.[34] In a Malone ACE, a tube goes through the abdominal wall to the appendix, or if available, to the colon. The bowel is then flushed daily.[35] Children as young as 6 years of age may administer this daily flush on their own.
If the affected portion of the lower intestine is restricted to the lower portion of the rectum, other surgical procedures may be performed, such as a posterior rectal myectomy.
The prognosis is good in 70% of cases. Chronic postoperative constipation is present in 7 to 8% of the operated cases. Postoperative enterocolitis, a severe manifestation, is present in the 10–20% of operated patients.
Epidemiology
According to a 1984 study conducted in Maryland, Hirschsprung's disease appears in 18.6 per 100,000 live births.[36] In Japan, it occurs at a similar rate of about one in 5,000 births (20 per 100,000).[37] It is more common in male than female (4.32:1) and in white rather than nonwhite.[38] Nine percent of the Hirschsprung cases were also diagnosed as having Down syndrome.[36] Most cases are diagnosed before the patient is 10 years of age.[7]
History
The first report of Hirschsprung's disease dates to 1691,[39] when it was described by Dutch anatomist Frederik Ruysch.[40] However, the disease is named after Harald Hirschsprung, the Danish physician who first described two infants who died of this disorder in 1888.[5][6]
Hirschsprung’s disease is a congenital disorder of the colon in which certain nerve cells, known as ganglion cells, are absent, causing chronic constipation.[41] The lack of ganglion cells is in the myenteric plexus (Auerbach's plexus), which is responsible for moving food in the intestine. A barium enema is the mainstay of diagnosis of Hirschsprung’s, though a rectal biopsy showing the lack of ganglion cells is the only certain method of diagnosis.
The first publication on an important genetic discovery of the disease was from Martucciello Giuseppe et al. in 1992. The authors described a case of a patient with total colonic aganglionosis associated with a 46, XX, del 10 (q11.21 q21.2) karyotype.[42] The major gene of Hirschsprung disease was identified in this chromosomal 10 region, it was the RET proto-oncogene.[43]
The usual treatment is "pull-through" surgery where the portion of the colon that does have nerve cells is pulled through and sewn over the part that lacks nerve cells.[44] For a long time, Hirschsprung’s was considered a multifactorial disorder, where a combination of nature and nurture was considered to be the cause. However, in August 1993, two articles by independent groups in Nature Genetics said that Hirschsprung’s disease could be mapped to a stretch of chromosome 10.[45][46]
This research also suggested that a single gene was responsible for the disorder. However, the researchers were unable to isolate it.
See also
- Achalasia
- Ileus, failure of peristaltic muscle activity in the gut
- Intestinal neuronal dysplasia
References
- ↑ 1.0 1.1 1.2 1.3 1.4 "Hirschsprung Disease". NORD (National Organization for Rare Disorders). 2017. Archived from the original on 2 June 2021. Retrieved 14 December 2017.
- ↑ 2.00 2.01 2.02 2.03 2.04 2.05 2.06 2.07 2.08 2.09 2.10 2.11 2.12 2.13 2.14 2.15 2.16 2.17 2.18 2.19 2.20 2.21 2.22 "Hirschsprung disease". Genetics Home Reference. August 2012. Archived from the original on 4 June 2021. Retrieved 14 December 2017.
- ↑ 3.00 3.01 3.02 3.03 3.04 3.05 3.06 3.07 3.08 3.09 3.10 "Hirschsprung's disease". Genetic and Rare Diseases Information Center (GARD) – an NCATS Program. 2017. Archived from the original on 24 November 2018. Retrieved 14 December 2017.
- ↑ Holschneider, Alexander Matthias; Puri, Prem (2007). Hirschsprung's Disease and Allied Disorders. Springer Science & Business Media. p. 1. ISBN 9783540339359. Archived from the original on 2017-12-14. Retrieved 2017-12-14.
- ↑ 5.0 5.1 "Hirschsprung's disease". www.whonamedit.com. Archived from the original on 24 March 2019. Retrieved 8 October 2019.
- ↑ 6.0 6.1 Hirschsprung, H. (1888). "Stuhlträgheit Neugeborener in Folge von Dilatation und Hypertrophie des Colons". Jahrbuch für Kinderheilkunde und physische Erziehung. Berlin. 27: 1–7.
- ↑ 7.0 7.1 7.2 7.3 7.4 7.5 Goldman, Lee. Goldman's Cecil Medicine (24th ed.). Philadelphia: Elsevier Saunders. p. 867. ISBN 978-1437727883.
- ↑ Kimura, Ken; Loening-Baucke, Vera (1999-11-01). "Failure to Pass Meconium: Diagnosing Neonatal Intestinal Obstruction". American Family Physician. 60 (7): 2043–50. ISSN 0002-838X. PMID 10569507. Archived from the original on 2019-08-18. Retrieved 2019-02-06.
- ↑ Martucciello G, Ceccherini I, Lerone M, Jasonni V (2000). "Pathogenesis of Hirschsprung's disease". Journal of Pediatric Surgery. 35 (7): 1017–1025. doi:10.1053/jpsu.2000.7763. PMID 10917288.
- ↑ Puri P, Shinkai T (2004). "Pathogenesis of Hirschsprung's disease and its variants: recent progress". Semin. Pediatr. Surg. 13 (1): 18–24. doi:10.1053/j.sempedsurg.2003.09.004. PMID 14765367. Archived from the original on 2021-08-29. Retrieved 2019-12-16.
- ↑ Garcia-Barcelo, Maria-Merce (2009). "Genome-wide association study identifies NRG1 as a susceptibility locus for Hirschsprung's disease". Proc. Natl. Acad. Sci. USA. 106 (8): 2694–2699. Bibcode:2009PNAS..106.2694G. doi:10.1073/pnas.0809630105. PMC 2650328. PMID 19196962.
- ↑ Tang, Clara (May 10, 2012). "Genome-wide copy number analysis uncovers a new HSCR gene: NRG3". PLoS Genet. 8 (5): e1002687. doi:10.1371/journal.pgen.1002687. PMC 3349728. PMID 22589734.
- ↑ Yang J, Duan S, Zhong R, Yin J, Pu J, Ke J, Lu X, Zou L, Zhang H, Zhu Z, Wang D, Xiao H, Guo A, Xia J, Miao X, Tang S, Wang G (2013). "Exome sequencing identified NRG3 as a novel susceptible gene of Hirschsprung's disease in a Chinese population". Mol. Neurobiol. 47 (3): 957–66. doi:10.1007/s12035-012-8392-4. PMID 23315268.
- ↑ Shin JG, Seo JY, Seo JM, Kim DY, Oh JT, Park KW, Kim HY, Kim JH, Shin HD (2019) Association analysis of NOX5 polymorphisms with Hirschsprung disease. J Pediatr Surg
- ↑ Online Mendelian Inheritance in Man (OMIM): 142623
- ↑ Mäkitie O, Heikkinen M, Kaitila I, Rintala R (2002). "Hirschsprung's disease in cartilage-hair hypoplasia has poor prognosis". J Pediatr Surg. 37 (11): 1585–8. doi:10.1053/jpsu.2002.36189. PMID 12407544.
- ↑ de Pontual L, Pelet A, Clement-Ziza M, Trochet D, Antonarakis SE, Attie-Bitach T, Beales PL, Blouin JL, Dastot-Le Moal F, Dollfus H, Goossens M, Katsanis N, Touraine R, Feingold J, Munnich A, Lyonnet S, Amiel J (2007). "Epistatic interactions with a common hypomorphic RET allele in syndromic Hirschsprung disease". Human Mutation. 28 (8): 790–6. doi:10.1002/humu.20517. PMID 17397038.
- ↑ Saunders CJ, Zhao W, Ardinger HH (2009). "Comprehensive ZEB2 gene analysis for Mowat-Wilson syndrome in a North American cohort: a suggested approach to molecular diagnostics". American Journal of Medical Genetics. 149A (11): 2527–31. doi:10.1002/ajmg.a.33067. PMID 19842203.
- ↑ Bonnard A, Zeidan S, Degas V, Viala J, Baumann C, Berrebi D, Perrusson O, El Ghoneimi A (2009). "Outcomes of Hirschsprung's disease associated with Mowat-Wilson syndrome". Journal of Pediatric Surgery. 44 (3): 587–91. doi:10.1016/j.jpedsurg.2008.10.066. PMID 19302864.
- ↑ Mueller C, Patel S, Irons M, Antshel K, Salen G, Tint GS, Bay C (2003). "Normal cognition and behavior in a Smith-Lemli-Opitz syndrome patient who presented with Hirschsprung disease". American Journal of Medical Genetics. 123A (1): 100–6. doi:10.1002/ajmg.a.20491. PMC 1201564. PMID 14556255.
- ↑ Flori E, Girodon E, Samama B, Becmeur F, Viville B, Girard-Lemaire F, Doray B, Schluth C, Marcellin L, Boehm N, Goossens M, Pingault V (2005). "Trisomy 7 mosaicism, maternal uniparental heterodisomy 7 and Hirschsprung's disease in a child with Silver-Russell syndrome". European Journal of Human Genetics. 13 (9): 1013–8. doi:10.1038/sj.ejhg.5201442. PMID 15915162.
- ↑ Torroglosa, Ana; Villalba-Benito, Leticia; Luzón-Toro, Berta; Fernández, Raquel María; Antiñolo, Guillermo; Borrego, Salud (January 2019). "Epigenetic Mechanisms in Hirschsprung Disease". International Journal of Molecular Sciences. 20 (13): 3123. doi:10.3390/ijms20133123. ISSN 1422-0067.
- ↑ Parisi MA, Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP (2002). Pagon RA, Bird TC, Dolan CR, Stephens K (eds.). "Hirschsprung Disease Overview". GeneReviews. PMID 20301612.
- ↑ Kays DW (1996). "Surgical conditions of the neonatal intestinal tract". Clinics in Perinatology. 23 (2): 353–75. doi:10.1016/S0095-5108(18)30246-X. PMID 8780909.
- ↑ Metallinos DL, Bowling AT, Rine J (Jun 1998). "A missense mutation in the endothelin-B receptor gene is associated with Lethal White Foal Syndrome: an equine version of Hirschsprung disease". Mamm. Genome. 9 (6): 426–31. doi:10.1007/s003359900790. PMID 9585428. Archived from the original on 2000-09-16.
- ↑ Dobbins WO, Bill AH (1965). "Diagnosis of Hirschsprung's Disease Excluded by Rectal Suction Biopsy". New England Journal of Medicine. 272 (19): 990–993. doi:10.1056/NEJM196505132721903. PMID 14279253.
- ↑ Eli Ehrenpreis (Oct 2003). Anal and rectal diseases explained. Remedica. pp. 15–. ISBN 978-1-901346-67-1. Archived from the original on 2017-04-22. Retrieved 2010-11-12.
- ↑ Martucciello G, Pini Prato A, Puri P, Holschneider AM, Meier-Ruge W, Jasonni V, Tovar JA, Grosfeld JL (2005). "Controversies concerning diagnostic guidelines for anomalies of the enteric nervous system: a report from the fourth International Symposium on Hirschsprung's disease and related neurocristopathies". J Pediatr Surg. 40 (10): 1527–31. doi:10.1016/j.jpedsurg.2005.07.053. PMID 16226977.
- ↑ Kim HJ, Kim AY, Lee CW, Yu CS, Kim JS, Kim PN, Lee MG, Ha HK (2008). "Hirschsprung disease and hypoganglionosis in adults: radiologic findings and differentiation". Radiology. 247 (2): 428–34. doi:10.1148/radiol.2472070182. PMID 18430875.
- ↑ Swenson O (1989). "My early experience with Hirschsprung's disease". J. Pediatr. Surg. 24 (8): 839–44, discussion 844–5. doi:10.1016/S0022-3468(89)80549-4. PMID 2671336.
- ↑ 31.0 31.1 "Hirschsprung disease". American Pediatric Surgical Association. Archived from the original on 9 December 2019. Retrieved 11 June 2019.
- ↑ W. Allan Walker (2004-07-01). Pediatric gastrointestinal disease: pathophysiology, diagnosis, management. PMPH-USA. pp. 2120–. ISBN 978-1-55009-240-0. Archived from the original on 2014-06-27. Retrieved 2010-11-12.
- ↑ Timothy R. Koch (2003). Colonic diseases. Humana Press. pp. 387–. ISBN 978-0-89603-961-2. Archived from the original on 2014-06-27. Retrieved 2010-11-12.
- ↑ Malone PS, Ransley PG, Kiely EM (1990). "Preliminary report: the antegrade continence enema". Lancet. 336 (8725): 1217–1218. doi:10.1016/0140-6736(90)92834-5. PMID 1978072.
- ↑ Walsh RA, Koyle MA, Waxman SW (2000). "The Malone ACE Procedure for Fecal Incontinence". Infections in Urology. 13 (4). Archived from the original on 2011-04-07. Retrieved 2012-01-04.
- ↑ 36.0 36.1 Goldberg EL (1984). "An epidemiological study of Hirschsprung's disease". Int J Epidemiol. 13 (4): 479–85. doi:10.1093/ije/13.4.479. PMID 6240474.
- ↑ Suita S, Taguchi T, Ieiri S, Nakatsuji T (2005). "Hirschsprung's disease in Japan: analysis of 3852 patients based on a nationwide survey in 30 years". Journal of Pediatric Surgery. 40 (1): 197–201, discussion 201–2. doi:10.1016/j.jpedsurg.2004.09.052. PMID 15868585.
- ↑ Colwell, Janice (2004). Fecal and Urinary Diversion Management. Mosby. p. 264. ISBN 978-0-323-02248-4.
- ↑ Hirschsprung's Disease and Allied Disorders. Berlin: Springer. 2007. ISBN 978-3-540-33934-2.
- ↑ "Hirschsprung Disease: Background, Pathophysiology, Epidemiology". 2017-01-08. Archived from the original on 2017-04-06. Retrieved 2017-04-05.
{{cite journal}}: Cite journal requires|journal=(help) - ↑ Worman S, Ganiats TG (1995). "Hirschsprung's disease: a cause of chronic constipation in children". Am Fam Physician. 51 (2): 487–94. PMID 7840044.
- ↑ Martucciello G; Bicocchi MP; Dodero P.; Lerone M.; Silengo Cirillo M; Puliti A; Gimelli G; Romeo G. (1992). "Total colonic aganglionosis associated with interstitial deletion of the long arm of chromosome 10". Pediatric Surgery International. 7 (4): 308–310. doi:10.1007/BF00183991.
- ↑ Romeo G, Ronchetto P, Luo Y, Barone V, Seri M, Ceccherini I, Pasini B, Bocciardi R, Lerone M, Kääriäinen H (1994). "Point mutations affecting the tyrosine kinase domain of the RET proto-oncogene in Hirschsprung's disease". Nature. 367 (6461): 377–378. Bibcode:1994Natur.367..377R. doi:10.1038/367377a0. PMID 8114938.
- ↑ (National Digestive Diseases Information Clearinghouse).
- ↑ Angrist M, Kauffman E, Slaugenhaupt SA, Matise TC, Puffenberger EG, Washington SS, Lipson A, Cass DT, Reyna T, Weeks DE (1993). "A gene for Hirschsprung disease (megacolon) in the pericentromeric region of human chromosome 10". Nat. Genet. 4 (4): 351–6. doi:10.1038/ng0893-351. PMID 8401581.
- ↑ Lyonnet S, Bolino A, Pelet A, Abel L, Nihoul-Fékété C, Briard ML, Mok-Siu V, Kaariainen H, Martucciello G, Lerone M, Puliti A, Luo Y, Weissenbach J, Devoto M, Munnich A, Romeo G (1993). "A gene for Hirschsprung disease maps to the proximal long arm of chromosome 10". Nat. Genet. 4 (4): 346–50. doi:10.1038/ng0893-346. PMID 8401580.
External links
| Classification | |
|---|---|
| External resources |
- Pages with script errors
- CS1 errors: missing periodical
- All articles with unsourced statements
- Articles with unsourced statements from August 2018
- Articles with invalid date parameter in template
- Articles with Curlie links
- Gastrointestinal tract disorders
- Congenital disorders of digestive system
- Rare diseases
- RTT