Henoch–Schönlein purpura
| Henoch-Schönlein purpura | |
|---|---|
| Other names: Schönlein–Henoch purpura,[1] IgA vasculitis | |
 | |
| Typical purpura on lower legs and buttocks | |
| Pronunciation | |
| Specialty | Rheumatology |
| Symptoms | Purpura, petechiae, joint pain, abdominal pain[2] |
| Complications | Kidney failure, intussusception[2] |
| Duration | 4 weeks[2] |
| Diagnostic method | Based on symptoms without evidence of low platelets[2] |
| Differential diagnosis | Hemolytic uremic syndrome, idiopathic thrombocytopenic purpura, disseminated intravascular coagulation, lupus, Rocky Mountain spotted fever[2] |
| Treatment | Supportive care, corticosteroids, plasma exchange, immunosuppressants[2] |
| Frequency | Rare[2] |
Henoch–Schönlein purpura (HSP), also known as IgA vasculitis, is inflammation of the small blood vessels of the skin, kidneys, joints, and gastrointestinal tract.[2] Less commonly the brain or lungs may be involved.[2] Symptoms include purpura (purple bruises) or petechiae (small red dots) in the skin, and potentially joint pain and abdominal pain.[2] The rash primarily affects the legs.[2] Complications may include kidney failure and intussusception.[2]
HSP is often preceded by an infection, such as a throat infection or gastroenteritis.[2] The underlying mechanism involves build up of immune complexes containing the antibody immunoglobulin A (IgA) in small blood vessels, resulting in inflammation.[2] The diagnosis is based on symptoms without evidence of low platelets.[2]
Treatment is generally with supportive care, unless the kidneys are involves.[2] Paracetamol (acetaminophen) and occasionally NSAIDs may be used for the pain.[2] If the kidneys are involved treatment may include corticosteroids, plasma exchange, immunosuppressants, and ACE inhibitors.[2] Most people get better within 4 weeks though there is a risk of reoccurrence.[2] In adults, complications are more frequent.[3]
The condition is rare.[2] Children are most commonly affected and in this population it occurring in about 15 per 100,000 per year.[2] It is named after the German physicians Johann Schonlein and Eduard Henoch though was described earlier by William Heberden in the early 1800s.[2]
Signs and symptoms
Purpura, arthritis, and abdominal pain are known as the "classic triad" of Henoch–Schönlein purpura.[4] Purpura occur in all cases, joint pains and arthritis in 80%, and abdominal pain in 62%. Some include gastrointestinal hemorrhage as a fourth criterion; this occurs in 33% of cases, sometimes, but not necessarily always, due to intussusception.[5] The purpura typically appear on the legs and buttocks, but may also be seen on the arms, face and trunk. The abdominal pain is colicky in character, and may be accompanied by nausea, vomiting, constipation or diarrhea. There may be blood or mucus in the stools.[6] The joints involved tend to be the ankles, knees, and elbows, but arthritis in the hands and feet is possible; the arthritis is nonerosive and hence causes no permanent deformity.[4] Forty percent have evidence of kidney involvement, mainly in the form of hematuria (blood in the urine), but only a quarter will have this in sufficient quantities to be noticeable without laboratory tests.[5] Problems in other organs, such as the central nervous system (brain and spinal cord) and lungs may occur, but is much less common than in the skin, bowel and kidneys.[7]
Of the 40% of people who develop kidney involvement, almost all have evidence (visible or on urinalysis) of blood in the urine. More than half also have proteinuria (protein in the urine), which in one eighth is severe enough to cause nephrotic syndrome (generalised swelling due to low protein content of the blood). While abnormalities on urinalysis may continue for a long time, only 1% of all HSP patients develop chronic kidney disease.[7] Hypertension (high blood pressure) may occur. Protein loss and high blood pressure, as well as the features on biopsy of the kidney if performed, may predict progression to advanced kidney disease. Adults are more likely than children to develop advanced kidney disease.[7][8]
-
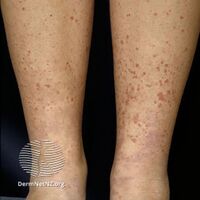
Henoch Schönlein purpura
-
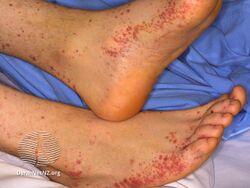
Henoch Schönlein purpura
-

Typical purpura on lower leg
-

More severe case of HSP on child's foot, leg, and arm
.jpg)
.jpg)


Cause
HSP can develop after infections with streptococci (β-haemolytic, Lancefield group A), hepatitis B, herpes simplex virus, parvovirus B19, Coxsackievirus, adenovirus, Helicobacter pylori,[7] measles, mumps, rubella, Mycoplasma and numerous others.[9] Drugs linked to HSP, usually as an idiosyncratic reaction, include the antibiotics vancomycin and cefuroxime, ACE inhibitors enalapril and captopril, anti-inflammatory agent diclofenac, as well as ranitidine and streptokinase. Several diseases have been reported to be associated with HSP, often without a causative link. Only in about 35% of cases can HSP be traced to any of these causes.[9]
The exact cause of HSP is unknown, but most of its features are due to the deposition of abnormal antibodies in the wall of blood vessels, leading to vasculitis. These antibodies are of the subclass IgA1 in polymers; it is uncertain whether the main cause is overproduction (in the digestive tract or the bone marrow) or decreased removal of abnormal IgA from the circulation.[9] It is suspected that abnormalities in the IgA1 molecule may provide an explanation for its abnormal behaviour in both HSP and the related condition IgA nephropathy. One of the characteristics of IgA1 (and IgD) is the presence of an 18 amino acid-long "hinge region" between complement-fixating regions 1 and 2. Of the amino acids, half is proline, while the others are mainly serine and threonine. The majority of the serines and the threonines have elaborate sugar chains, connected through oxygen atoms (O-glycosylation). This process is thought to stabilise the IgA molecule and make it less prone to proteolysis. The first sugar is always N-acetyl-galactosamine (GalNAc), followed by other galactoses and sialic acid. In HSP and IgAN, these sugar chains appear to be deficient. The exact reason for these abnormalities is not known.[7][9]
Pathophysiology
Henoch–Schönlein purpura is a small-vessel vasculitis in which complexes of immunoglobulin A (IgA) and complement component 3 (C3) are deposited on arterioles, capillaries, and venules (hence it is a type III hypersensitivity reaction). As with IgA nephropathy, serum levels of IgA are high in HSP and there are identical findings on renal biopsy; however, IgA nephropathy has a predilection for young adults while HSP is more predominant among children. Further, IgA nephropathy typically only affects the kidneys while HSP is a systemic disease. HSP involves the skin and connective tissues, scrotum, joints, gastrointestinal tract and kidneys.[9] The genetic basis remains unclear except for involvement of the human leukocyte antigen region of the genome.[10] It is hypothesized to involve autoimmunity triggered by infections, particularly Staphylococcus aureus, Mycoplasma and adenovirus.[citation needed]
Diagnosis


The diagnosis is based on the combination of the symptoms, as very few other diseases cause the same symptoms together. Blood tests may show elevated creatinine and urea levels (in kidney involvement), raised IgA levels (in about 50%[9]), and raised C-reactive protein (CRP) or erythrocyte sedimentation rate (ESR) results; none are specific for Henoch–Schönlein purpura. The platelet count may be raised, and distinguishes it from diseases where low platelets are the cause of the purpura, such as idiopathic thrombocytopenic purpura and thrombotic thrombocytopenic purpura.[4]
If there is doubt about the cause of the skin lesions, a biopsy of the skin may be performed to distinguish the purpura from other diseases that cause it, such as vasculitis due to cryoglobulinemia; on microscopy the appearances are of a hypersensitivity vasculitis, and immunofluorescence demonstrates IgA and C3 (a protein of the complement system) in the blood vessel wall.[4] However, overall serum complement levels are normal.
On the basis of symptoms, it is possible to distinguish HSP from hypersensitivity vasculitis (HV). In a series comparing 85 HSP patients with 93 HV patients, five symptoms were found to be indicative of HSP: palpable purpura, abdominal angina, digestive tract hemorrhage (not due to intussussception), hematuria and age less than 20. The presence of three or more of these indicators has an 87% sensitivity for predicting HSP.[11]
Biopsy of the kidney may be performed both to establish the diagnosis or to assess the severity of already suspected kidney disease. The main findings on kidney biopsy are increased cells and Ig deposition in the mesangium (part of the glomerulus, where blood is filtered), white blood cells, and the development of crescents. The changes are indistinguishable from those observed in IgA nephropathy.[9]
Classification
Multiple standards exist for defining Henoch–Schönlein purpura, including the 1990 American College of Rheumatology (ACR) classification[12][13] and the 1994 Chapel Hill Consensus Conference (CHCC).[14] Some have reported the ACR criteria to be more sensitive than those of the CHCC.[15]
More recent classifications, the 2006 European League Against Rheumatism (EULAR) and Pediatric Rheumatology Society (PReS) classification, include palpable purpura as a mandatory criterion, together with at least one of the following findings: diffuse abdominal pain, predominant IgA deposition (confirmed on skin biopsy), acute arthritis in any joint, and renal involvement (as evidenced by the presence of blood and/or protein in the urine).[16]
Differential diagnosis
Henoch–Schönlein purpura may present with an atypical manifestation, which can be confused with papular urticaria, systemic lupus erythematosus, meningococcemia, dermatitis herpetiformis, and acute hemorrhagic edema of infancy.[17]
Treatment
As of 2017, the optimal way to treat Henoch–Schönlein purpura remains controversial.[18] Analgesics may be needed for the abdominal and joint pains. Wound care is warranted if the skin death and ulcerations occur.[18] It is uncertain as to whether HSP needs treatment beyond controlling the symptoms. Most people do not receive therapy because of the high spontaneous recovery rate. Experts disagree on whether to routinely use corticosteroids as treatment for HSP.[18] However, if they are given early in the disease episode, the duration of symptoms may be shortened, and abdominal pain can improve significantly.[18] Moreover, the chance of severe kidney problems may be reduced.[19] A systematic review did not find any evidence that steroid treatment (prednisone) is effective at decreasing the likelihood of developing long-term kidney disease.[20]
Evidence of worsening kidney damage would normally prompt a kidney biopsy. Treatment may be indicated on the basis of the appearance of the biopsy sample; various treatments may be used, ranging from steroids by mouth to a combination of intravenous methylprednisolone (steroid), cyclophosphamide and dipyridamole followed by prednisone. Other regimens include steroids/azathioprine, and steroids/cyclophosphamide (with or without heparin and warfarin). Intravenous immunoglobulin (IVIG) is occasionally used.[9]
There is no good evidence that treating children who have HSP with antiplatelet agent prevents persistent kidney disease.[20] There is also no evidence that treating children or adults with cyclophosphamide prevents severe kidney disease.[20] Heparin treatment is not justified.[20]
Prognosis
Overall prognosis is good in most patients, with one study showing recovery occurring in 94% and 89% of children and adults, respectively (some having needed treatment).[21] In children under ten, the condition recurs in about a third of all cases, usually within the four months of the initial attack.[5] Recurrence is more common in older children and adults.[7]
Kidneys
In adults, kidney involvement progresses to end-stage kidney disease (ESKD) more often than in children. In a UK series of 37 patients, 10 (27%) developed advanced kidney disease. Proteinuria, hypertension at presentation, and pathology features (crescentic changes, interstitial fibrosis and tubular atrophy) predicted progression.[8] About 20% of children that exhibit nephrotic or nephritic features experience long permanent renal impairment.[22]
The findings on renal biopsy correlate with the severity of symptoms: those with asymptomatic hematuria may only have focal mesangial proliferation while those with proteinuria may have marked cellular proliferation or even crescent formation. The number of crescentic glomeruli is an important prognostic factor in determining whether the patient will develop chronic renal disease.[7]
In ESKD, some eventually need hemodialysis or equivalent renal replacement therapy (RRT). If a kidney transplant is found for a patient on RRT, the disease will recur in the graft (transplanted kidney) in about 35% of cases, and in 11%, the graft will fail completely (requiring resumption of the RRT and a further transplant).[9]
Epidemiology
HSP occurs more often in children than in adults,[21] and usually follows an upper respiratory tract infection. Half of affected patients are below the age of six, and 90% are under ten. It occurs about twice as often in boys as in girls.[7] The incidence of HSP in children is about 20 per 100,000 children per year, making it the most common vasculitis in children.[23]
Cases of HSP may occur anytime throughout the year, but some studies have found that fewer cases occur during the summer months.[24]
History
The disease is named after Eduard Heinrich Henoch (1820–1910), a German pediatrician (nephew of Moritz Heinrich Romberg) and his teacher Johann Lukas Schönlein (1793–1864), who described it in the 1860s. Schönlein associated the purpura and arthritis, and Henoch the purpura and gastrointestinal involvement. The English physician William Heberden (1710–1801) and the dermatologist Robert Willan (1757–1812) had already described the disease in 1802 and 1808, respectively, but the name Heberden–Willan disease has fallen into disuse. William Osler was the first to recognise the underlying allergic mechanism of HSP.[25]
See also
References
- ↑ Rapini RP, Bolognia JL, Jorizzo JL (2007). Dermatology. St. Louis: Mosby. ISBN 978-1-4160-2999-1.
- ↑ 2.00 2.01 2.02 2.03 2.04 2.05 2.06 2.07 2.08 2.09 2.10 2.11 2.12 2.13 2.14 2.15 2.16 2.17 2.18 2.19 2.20 2.21 Roache-Robinson, P; Hotwagner, DT (January 2020). "Henoch Schonlein Purpura". PMID 30725937.
{{cite journal}}: Cite journal requires|journal=(help) - ↑ "Treatment Challenges, Uncertainty Abound with IgA Vasculitis". The Rheumatologist. 2016. Archived from the original on 2018-12-16. Retrieved 2017-08-20.
- ↑ 4.0 4.1 4.2 4.3 Kraft DM, Mckee D, Scott C (1998). "Henoch-Schönlein purpura: a review". American Family Physician. 58 (2): 405–8, 411. PMID 9713395. Archived from the original on 2011-06-06. Retrieved 2007-12-15.
- ↑ 5.0 5.1 5.2 Saulsbury FT (1999). "Henoch-Schönlein purpura in children. Report of 100 patients and review of the literature". Medicine (Baltimore). 78 (6): 395–409. doi:10.1097/00005792-199911000-00005. PMID 10575422.
- ↑ Fauci AS (1987). "269:The Vasculitis Syndromes". In Braunwald E, Isselbacher KJ, Petersdorf RG, Wilson JD, Martin JB, Fauci AS (eds.). Harrison's Book of Internal Medicine. Vol. 2 (11th ed.). McGraw Hill. p. 1441. ISBN 978-0-07-079454-2.
- ↑ 7.0 7.1 7.2 7.3 7.4 7.5 7.6 7.7 Saulsbury FT (2001). "Henoch-Schönlein purpura". Current Opinion in Rheumatology. 13 (1): 35–40. doi:10.1097/00002281-200101000-00006. PMID 11148713.
- ↑ 8.0 8.1 Shrestha S, Sumingan N, Tan J, et al. (2006). "Henoch Schönlein purpura with nephritis in adults: adverse prognostic indicators in a UK population". QJM. 99 (4): 253–65. doi:10.1093/qjmed/hcl034. PMID 16565522. Archived from the original on 2009-06-29. Retrieved 2007-12-16.
- ↑ 9.0 9.1 9.2 9.3 9.4 9.5 9.6 9.7 9.8 Rai A, Nast C, Adler S (1 December 1999). "Henoch-Schönlein purpura nephritis". Journal of the American Society of Nephrology. 10 (12): 2637–44. PMID 10589705. Archived from the original on 3 June 2009. Retrieved 16 December 2007.
- ↑ López-Mejías, Raquel; Castañeda, Santos; Genre, Fernanda; Remuzgo-Martínez, Sara; Carmona, F. David; Llorca, Javier; Blanco, Ricardo; Martín, Javier; González-Gay, Miguel A. (March 2018). "Genetics of immunoglobulin-A vasculitis (Henoch-Schönlein purpura): An updated review". Autoimmunity Reviews. 17 (3): 301–315. doi:10.1016/j.autrev.2017.11.024. PMID 29353097.
- ↑ Michel BA, Hunder GG, Bloch DA, Calabrese LH (1992). "Hypersensitivity vasculitis and Henoch-Schönlein purpura: a comparison between the 2 disorders". Journal of Rheumatology. 19 (5): 721–8. PMID 1613701.
- ↑ Mills JA, Michel BA, Bloch DA, et al. (1990). "The American College of Rheumatology 1990 criteria for the classification of Henoch-Schönlein purpura". Arthritis and Rheumatism. 33 (8): 1114–21. doi:10.1002/art.1780330809. PMID 2202310.
- ↑ American College of Rheumatology. "1990 criteria for the classification of Henoch-Schönlein purpura". Archived from the original on 2016-03-03. Retrieved 2007-12-15.
- ↑ Jennette JC, Falk RJ, Andrassy K, et al. (1994). "Nomenclature of systemic vasculitides. Proposal of an international consensus conference". Arthritis and Rheumatism. 37 (2): 187–92. doi:10.1002/art.1780370206. PMID 8129773.
- ↑ Murali NS, George R, John GT, et al. (2002). "Problems of classification of Henoch Schonlein purpura: an Indian perspective". Clinical and Experimental Dermatology. 27 (4): 260–3. doi:10.1046/j.1365-2230.2002.01063.x. PMID 12139664. S2CID 45849349.
- ↑ Ozen S, Ruperto N, Dillon MJ, et al. (July 2006). "EULAR/PReS endorsed consensus criteria* for the classification of childhood vasculitides". Annals of the Rheumatic Diseases. 65 (7): 936–41. doi:10.1136/ard.2005.046300. PMC 1798210. PMID 16322081.
- ↑ Lawee D (2008). "Atypical clinical course of Henoch-Schonlein purpura". Can Fam Physician (Review. Case Reports.). 54 (8): 1117–20. PMC 2515239. PMID 18697972.
- ↑ 18.0 18.1 18.2 18.3 Hetland, LE; Susrud, KS; Lindahl, KH; Bygum, A (November 2017). "Henoch-Schönlein Purpura: A Literature Review". Acta Dermato-venereologica (Review). 97 (10): 1160–66. doi:10.2340/00015555-2733. PMID 28654132.
- ↑ Weiss PF, Feinstein JA, Luan X, Burnham JM, Feudtner C (2007). "Effects of corticosteroid on Henoch-Schönlein purpura: a systematic review". Pediatrics. 120 (5): 1079–87. doi:10.1542/peds.2007-0667. PMC 3525094. PMID 17974746.
- ↑ 20.0 20.1 20.2 20.3 Hahn, Deirdre; Hodson, Elisabeth M.; Willis, Narelle S.; Craig, Jonathan C. (2015-08-07). "Interventions for preventing and treating kidney disease in Henoch-Schönlein Purpura (HSP)". The Cochrane Database of Systematic Reviews (8): CD005128. doi:10.1002/14651858.CD005128.pub3. ISSN 1469-493X. PMID 26258874.
- ↑ 21.0 21.1 Blanco R, Martínez-Taboada VM, Rodríguez-Valverde V, García-Fuentes M, González-Gay MA (1997). "Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome". Arthritis and Rheumatism. 40 (5): 859–64. doi:10.1002/art.1780400513. PMID 9153547.
- ↑ Watson, L; Richardson, AR; Holt, RC; Jones, CA; Beresford, MW (January 2012). "Henoch schonlein purpura--a 5-year review and proposed pathway". PLOS ONE. 7 (1): e29512. Bibcode:2012PLoSO...729512W. doi:10.1371/journal.pone.0029512. PMC 3250434. PMID 22235302.
- ↑ Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR (2002). "Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins". Lancet. 360 (9341): 1197–202. doi:10.1016/S0140-6736(02)11279-7. PMID 12401245. S2CID 25018798.
- ↑ Saulsbury FT (2002). "Epidemiology of Henoch-Schönlein purpura". Cleveland Clinic Journal of Medicine. 69 Suppl 2: SII87–9. doi:10.3949/ccjm.69.suppl_2.sii87. PMID 12086273. Archived from the original on 2020-03-27. Retrieved 2012-08-26.
- ↑ Schönlein-Henoch purpura at Who Named It?
External links
| Classification | |
|---|---|
| External resources |
- Pages with script errors
- CS1 errors: missing periodical
- CS1: long volume value
- All articles with unsourced statements
- Articles with unsourced statements from December 2019
- Articles with invalid date parameter in template
- Good articles
- Rheumatology
- Pediatrics
- Nephrology
- Autoimmune diseases
- Ailments of unknown cause
- Vascular-related cutaneous conditions
- RTT