Greig cephalopolysyndactyly syndrome
| Greig cephalopolysyndactyly syndrome | |
|---|---|
 | |
| Patient with Greig cephalopolysyndactyly syndrome showing hypertelorism and macrocephaly. | |
Greig cephalopolysyndactyly syndrome is a disorder that affects development of the limbs, head, and face. The features of this syndrome are highly variable, ranging from very mild to severe. People with this condition typically have one or more extra fingers or toes (polydactyly) or an abnormally wide thumb or big toe (hallux).[1]
Signs and symptoms

The skin between the fingers and toes may be fused (cutaneous syndactyly). This disorder is also characterized by widely spaced eyes (ocular hypertelorism), an abnormally large head size (macrocephaly), and a high, prominent forehead. Rarely, affected individuals may have more serious medical problems including seizures and developmental delay.[1]
Pathophysiology

Greig cephalopolysyndactyly syndrome is a chromosomal condition related to chromosome 7. Mutations in the GLI3 gene cause Greig cephalopolysyndactyly syndrome. The GLI3 gene provides instructions for making a protein that controls gene expression, which is a process that regulates whether genes are turned on or off in particular cells. By interacting with certain genes at specific times during development, the GLI3 protein plays a role in the normal shaping (patterning) of many organs and tissues before birth.[1]
Different genetic changes involving the Gli3 gene can cause Greig cephalopolysyndactyly syndrome. In some cases, the condition results from a chromosomal abnormality, such as a large deletion or translocation of genetic material, in the region of chromosome 7 that contains the GLI3 gene. In other cases, a mutation in the GLI3 gene itself is responsible for the disorder. Each of these genetic changes prevents one copy of the gene in each cell from producing any functional protein. It remains unclear how a reduced amount of this protein disrupts early development and causes the characteristic features of Greig cephalopolysyndactyly syndrome.[1]
This condition is inherited in an autosomal dominant pattern, which means the defective gene is located on an autosome, and only one copy of the defective GLI3 gene is sufficient to cause the disorder. In cases of dominant inheritance, an affected person inherits the genetic mutation or chromosomal abnormality from one affected parent.[1]
Rare instances of this disorder are sporadic, and occur in people with no history of the condition in their family.[1]
Diagnosis
The disease is not easily definable. The main form of diagnosis is presumptive, if the person has the usual triad of preaxial polydactyly with cutaneous syndactyly of at least one limb, macrocephaly, and hypertelorism. However, a definitive diagnosis can be made if there is a phenotype that is caused by a Gcps and a Gli3 gene mutation. It can also be made if the person has a family member or relative with the disease. Possibly, antenatal ultrasound can detect macrocephaly, and a high-resolution ultrasound can detect polydactyly and syndactyly. There are a lot of differential diagnoses, including over 100 syndromes and disorders that can cause polydactyly. However, there are a few diseases with an overlap that is significant. These diseases include: acrocallosal syndrome, carpenter syndrome, and Gorlin syndrome. [1]
-
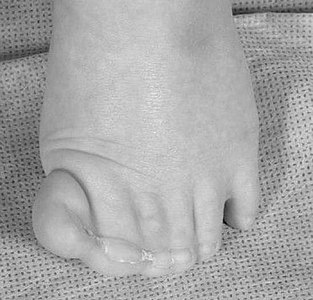
The foot of the patient with Greig cephalopolysyndactyly shows a partially duplicated hallux with cutaneous syndactyly of several digits.
-

Finger with syndactyly of many digits caused by Greig cephalopolysyndactyly.


Management
The main treatment is surgery to fix the abnormalities in the limbs, like syndactyly. It is less important to repair the feet surgically, as it can cause complications, and it is not aesthetically important compared to the hands. If there is polydactyly with an extra digit that is fully functional, then the digit can stay. Some people may have intellectual disability, though this condition usually causes only mild impairment. Developmental assistance and early specialist intervention should be offered in the case of intellectual disability. People should be tested as an individual and be given proper assistance.[1]
Prognosis
The outlook is usually good in the usual case of GCPS. Mostly, the biggest problem associated with the condition in mild intellectual impairment.[citation needed]
Eponym
The condition is named for David Middleton Greig FRSE.[2]
References
External links
- Greig cephalopolysyndactyly syndrome at NLM Genetics Home Reference
- GeneReview/NIH/UW entry on Greig Cephalopolysyndactyly Syndrome Archived 2020-11-26 at the Wayback Machine
| Classification | |
|---|---|
| External resources |
- Pages with script errors
- Articles with hatnote templates targeting a nonexistent page
- Missing redirects
- All articles with unsourced statements
- Articles with unsourced statements from October 2020
- Articles with invalid date parameter in template
- Webarchive template wayback links
- Congenital disorders of musculoskeletal system
- Transcription factor deficiencies
- Autosomal dominant disorders
- Syndromes with dysmelia
- Syndromes affecting head size
- Rare syndromes