Ewing sarcoma
| Ewing sarcoma | |
|---|---|
| Other names: Peripheral primitive neuroectodermal tumor (not recommended), Askin tumor (not recommended),[1] Ewing sarcoma family of tumors (previous term)[2] | |
  | |
| Top: Ewing sarcoma of the thigh bone on a child's CT scan Bottom: Extraskeletal Ewing sarcoma of the hard palate | |
| Pronunciation | |
| Specialty | Oncology |
| Symptoms | Swelling and pain near the tumor[1] |
| Complications | Spread, broken bone[3] |
| Usual onset | 10 to 20 years old[1] |
| Types | Ewing sarcoma of bone, extraskeletal Ewing tumor[1] |
| Causes | Unknown[1] |
| Diagnostic method | Medical imaging, tissue biopsy[3] |
| Differential diagnosis | Osteosarcoma, neuroblastoma, osteomyelitis[4] |
| Treatment | Chemotherapy, radiation therapy, surgery, stem cell transplant[3] |
| Prognosis | Five year survival ~ 70%[5] |
| Frequency | 1 per million people (US)[5] |
Ewing sarcoma (EwS) is a type of cancer that can form in bone or in soft tissue.[1] Symptoms may include swelling and pain at the site of the tumor, fever, or a bone fracture.[3] The most common areas affected are the shafts of long bones of the legs, the pelvis, and the chest wall.[5] In soft tissue, it can affect various parts of the body.[6] In about 25% of cases, the cancer has already spread to other parts of the body at the time of diagnosis.[7]
The cause of a Ewing sarcoma is unknown.[1] Most cases appear to occur randomly.[1] It is a type of small round cell sarcoma.[1] Around 85% of cases are positive for CD99, a cell-surface glycoprotein.[1] The underlying mechanism often involves a genetic rearrangement, most commonly between EWSR1 and FLII.[8] Diagnosis is based on biopsy of the tumor; microscopic appearance and genetic confirmation.[1] Other conditions that may appear similar include osteosarcoma, neuroblastoma, osteomyelitis, rickets, and rhabdomyosarcoma.[4]
Treatment often includes chemotherapy, radiation therapy, surgery, and stem cell transplant.[3] Targeted therapy and immunotherapy are being studied.[2] Five year survival is about 70%.[5] A number of factors; however, affect this estimate.[5]
Ewing sarcoma most frequently occurs in teenagers and young adults, but one in five may be in older people.[8] In the United States about one in a million people per year are affected.[5] It represents 2% of childhood cancers.[2] Caucasians are affected more often than African Americans or Asians.[5] Males are affected more often than females.[5] James Ewing first described the cancer in 1920 and it was later named after him by Ernest Amory Codman.[9]
Types
There are generally two types; most are Ewing sarcoma of bone, and around 12% are extraskeletal Ewing sarcoma that forms in soft tissue.[1]
Signs and symptoms

Features include most commonly pain and a lump at the site of the tumor, usually in a person under the age of 20 years.[1] There may be fever or an unexpected broken bone.[1]
It can occur anywhere in the body, but most commonly in the pelvis and the long tubular bones, especially around the growth plates.[1] The diaphyses of the thigh bone are the most common sites, followed by the tibia and the upper arm bone.[citation needed] Around 25% have spread at presentation.[1] In soft tissue, it can affect a wide distribution of the body.[6] Rarely, it can develop in the cervix, vagina and vulva.[10]
Cause
Most cases appear to occur randomly.[1] Sometimes there has been a germline mutation.[1]
Genetics
Genetic exchange between chromosomes can cause cells to become cancerous. Most cases of Ewing sarcoma (85%) are the result of a translocation between chromosomes 11 and 22, which fuses the EWSR1 gene of chromosome 22 to the FLI1 gene of chromosome 11.[11]
Diagnosis
Initial tests include medical imaging.[1] Diagnosis is by biopsy and is based on histomorphologic findings and immunohistochemistry, followed by genetic confirmation.[1] Either open biopsy, or core needle biopsy with a CT/MRI scan is required for accurate sarcoma diagnosis[3]
Medical imaging
On conventional radiographs, the most common osseous presentation is a permeative lytic lesion with periosteal reaction.[1] The classic description of lamellated or "onion-skin" type periosteal reaction is often associated with this lesion.[1] In addition, Magnetic resonance imaging (MRI), Computed axial tomography (CT) and PET scan can help fully define the Ewing sarcoma and assess any spread.[1]
MRI will show the full bony and soft tissue extent and relate the tumor to other nearby anatomic structures (e.g. vessels). Gadolinium contrast is not necessary as it does not give additional information over noncontrast studies, though some current researchers argue that dynamic, contrast-enhanced MRI may help determine the amount of necrosis within the tumor, thus help in determining response to treatment prior to surgery. Computed axial tomography (CT) can also be used to define the extraosseous extent of the tumor, especially in the skull, spine, ribs, and pelvis. Both CT and MRI can be used to follow response to radiation and/or chemotherapy. Bone scintigraphy can also be used to follow tumor response to therapy. In the group of malignant small round cell tumors that includes Ewing sarcoma, bone lymphoma, and small cell osteosarcoma, the cortex may appear almost normal radiographically, while permeative growth occurs throughout the Haversian channels. These tumors may be accompanied by a large soft-tissue mass while almost no bone destruction is visible. The radiographs frequently do not shown any signs of cortical destruction. Radiographically, Ewing's sarcoma presents as "moth-eaten" destructive radiolucencies of the medulla and erosion of the cortex with expansion.[citation needed]
-

X-ray of a child with Ewing sarcoma of the tibia
-

MRI slice showing Ewing sarcoma of the left hip (white area shown right)
-
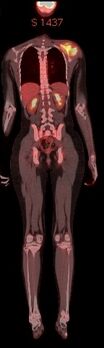
PET-CT scan: increased FDG uptake in the soft tissue mass of left scapula.
-
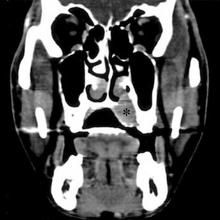
CT scan face- extraskeletal Ewing sarcoma hard palate




Histopathology
There are typically many small cells of the same size and appearance, with round nuclei.[3] Around half have extensive glycogen deposits, demonstrated with positive PAS staining.[3] Around 85% of Ewing sarcomas are positive for CD99, a cell-surface glycoprotein.[1] However, CD99 also occurs in some normal tissue and in other round cell sarcomas, as well as in lymphoblastic lymphoma and leukemia.[3] There may be keratin expression.[1]
-

Micrograph of metastatic Ewing sarcoma (right of image) in normal lung (left of image). PAS stain
-

Micrograph of a metastatic Ewing sarcoma with the characteristic cytoplasmic clearing on H&E staining, which was showing to be PAS positive


Differential diagnosis
The differential diagnosis includes other small-blue-round-cell tumors, which includes lymphoma, alveolar rhabdomyosarcoma, and desmoplastic small round cell tumor, among others.[3] Other conditions that may appear similar include osteosarcoma, neuroblastoma, osteomyelitis and rickets.[4]
Treatment
Almost all people receive multidrug chemotherapy (most often vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide), as well as local disease control with surgery and/or radiation.[3] An aggressive approach is necessary because almost all people with apparently localized disease at the time of diagnosis actually have asymptomatic metastatic disease (microscopic spread).[3]
The surgical resection may involve limb salvage or amputation.[3] Proton beam therapy delivers an equally effective dose to the sarcoma with less damage to the surrounding normal tissue compared to photon radiation, particularly for children.[3]
Complete excision at the time of biopsy may be performed if malignancy is confirmed at the time it is examined.[citation needed] Treatment lengths vary depending on location and stage of the disease at diagnosis. Radical chemotherapy may be as short as six treatments at three-week cycles, but most people undergo chemotherapy for 6–12 months and radiation therapy for 5–8 weeks.[citation needed] Radiotherapy has been used for localized disease. The tumor has a unique property of being highly sensitive to radiation, sometimes acknowledged by the phrase "melting like snow", but the main drawback is that it recurs dramatically after some time.[citation needed] Antisense oligodeoxynucleotides have been proposed as possible treatment by down-regulating the expression of the oncogenic fusion protein associated with the development of Ewing sarcoma resulting from the EWS-ETS gene translocation.[12][13] In addition, the synthetic retinoid derivative fenretinide (4-hydroxy(phenyl)retinamide) has been reported to induce high levels of cell death in Ewing sarcoma cell lines in vitro and to delay growth of xenografts in in vivo mouse models.[14][15]
Outcomes
Most often, metastases locate to the lungs, bone and/or bone marrow.[3] If found to be initially localised, 30% to 40% recur.[3] This increases to 60% to 80% if there is already spread at diagnosis.[3]
Five-year survival varies with diagnosis, type of treatment and where treated.[3] Prior to the use of multi-drug chemotherapy, long-term survival was less than 10%. The development of multi-disciplinary therapy with chemotherapy, irradiation, and surgery has increased current long-term survival rates in most clinical centers to greater than 50%.[16] However, some sources state it is 25–30%.[17]
Retrospective research showed that two chemokine receptors, CXCR4 and CXCR7, can be used as molecular prognosis factors. People who express low levels of both chemokine receptors have the highest odds of long-term survival with >90% survival at five years post-diagnosis versus <30% survival at five years for patients with very high expression levels of both receptors.[18] A recent study also suggested a role for SOX2 as an independent prognostic biomarker that can be used to identify patients at high risk for tumor relapse.[19]
Ewing sarcoma has poor prognosis and outcome in ~70% of initial diagnoses and 10–15% of relapses.[20]
Epidemiology
Ewing sarcoma is the second commonest bone cancer in children, with 80% of cases diagnosed before the age of 20 years.[1] Males are affected more than females at a M:F ratio of 1.4:1.[1] Around 25% of people diagnosed with Ewing sarcoma have spread.[1]
Extraskeletal Ewing sarcoma is 10 times less common than Ewing sarcoma of bone and occurs in around 1.4 per million people, with a greater likelihood in under 5-year olds and over 35-year olds.[6][21] There does not appear to be any association with ethnicity or gender.[6]
Ewing sarcomas represent 16% of primary bone sarcomas.[11] It is less common in people of African descent.[1]
In the United States, they are most common in the second decade of life,[11] with a rate of 0.3 cases per million in children under 3 years of age, and as high as 4.6 cases per million in adolescents aged 15–19 years. Internationally, the annual incidence rate averages less than 2 cases per million children.[22]
History
Ewing sarcoma is named for American pathologist James Ewing (1866-1943), who first described the tumor as a 'diffuse endothelium of bone' in 1920; it was later named for him by Ernest Amory Codman.[9] The concept of extraskeletal Ewing sarcoma was introduced in 1980.[3] However, the first reported case was in 1969 by Melvin Tefft.[23] The terms primitive neuroectodermal tumors and Askin tumors were later abandoned as distinct entities and the 2013 WHO classification of sarcomas uniformly defined ‘Ewing sarcoma’ as comprising both as they shared the phenotypic spectrum of Ewing sarcoma.[3] Previously referred to as Ewing-like sarcomas, round cell sarcoma with EWSR1- non-ETS fusions, CIC-rearranged sarcoma, and sarcoma with BCOR genetic alterations, because they appear similar, were later found not have similar genetic backgrounds, hence are termed "related entities".[3]
References
- ↑ 1.00 1.01 1.02 1.03 1.04 1.05 1.06 1.07 1.08 1.09 1.10 1.11 1.12 1.13 1.14 1.15 1.16 1.17 1.18 1.19 1.20 1.21 1.22 1.23 1.24 1.25 1.26 1.27 1.28 WHO Classification of Tumours Editorial Board, ed. (2020). "2. Undifferentiated small round cell sarcoma of bone and soft tissue: Ewing sarcoma". Soft Tissue and Bone Tumours: WHO Classification of Tumours. Vol. 3 (5th ed.). Lyon (France): International Agency for Research on Cancer. pp. 323–325. ISBN 978-92-832-4503-2. Archived from the original on 2021-06-13. Retrieved 2021-05-09.
- ↑ 2.0 2.1 2.2 "Ewing sarcoma treatment". National Cancer Institute. 9 May 2021. Archived from the original on 9 May 2021. Retrieved 9 May 2021.
- ↑ 3.00 3.01 3.02 3.03 3.04 3.05 3.06 3.07 3.08 3.09 3.10 3.11 3.12 3.13 3.14 3.15 3.16 3.17 3.18 3.19 3.20 Zöllner, SK; Amatruda, JF; Bauer, S; Collaud, S; de Álava, E; DuBois, SG; Hardes, J; Hartmann, W; Kovar, H; Metzler, M; Shulman, DS; Streitbürger, A; Timmermann, B; Toretsky, JA; Uhlenbruch, Y; Vieth, V; Grünewald, TGP; Dirksen, U (14 April 2021). "Ewing Sarcoma-Diagnosis, Treatment, Clinical Challenges and Future Perspectives". Journal of clinical medicine. 10 (8). doi:10.3390/jcm10081685. PMID 33919988. Archived from the original on 16 June 2022. Retrieved 23 June 2022.
- ↑ 4.0 4.1 4.2 Toretsky, Jeffrey A .T. (3 March 2021). "Ewing Sarcoma Differential Diagnoses". emedicine.medscape.com. Archived from the original on 24 September 2017. Retrieved 22 June 2022.
- ↑ 5.0 5.1 5.2 5.3 5.4 5.5 5.6 5.7 "Ewing Sarcoma Treatment". National Cancer Institute. 31 January 2019. Archived from the original on 8 January 2020. Retrieved 4 February 2019.
- ↑ 6.0 6.1 6.2 6.3 Abboud, A; Masrouha, K; Saliba, M; Haidar, R; Saab, R; Khoury, N; Tawil, A; Saghieh, S (May 2021). "Extraskeletal Ewing sarcoma: Diagnosis, management and prognosis". Oncology letters. 21 (5): 354. doi:10.3892/ol.2021.12615. PMID 33747211. Archived from the original on 2022-06-24. Retrieved 2022-06-23.
- ↑ Eaton, Bree R.; Claude, Line; Indelicato, Daniel J.; Vatner, Ralph; Yeh, Brian; Schwarz, Rudolf; Laack, Nadia (May 2021). "Ewing sarcoma". Pediatric Blood & Cancer. 68 Suppl 2: e28355. doi:10.1002/pbc.28355. ISSN 1545-5017. PMID 33818887. Archived from the original on 2022-06-10. Retrieved 2022-06-12.
- ↑ 8.0 8.1 Kontoyiannis, Dimitrios P. (2020). "192. Malignant tumors of bone, sarcomas, and other soft tissue neoplasms". In Goldman, Lee; Schafer, Andrew I. (eds.). Goldman-Cecil Medicine. Vol. 1 (26th ed.). Philadelphia: Elsevier. pp. 1341–1342. ISBN 978-0-323-55087-1. Archived from the original on 2022-06-24. Retrieved 2022-06-22.
- ↑ 9.0 9.1 Mostofi, Seyed B. (2004). Who's Who in Orthopedics. London: Springer. p. 101. ISBN 1-85233-786-9. Archived from the original on 2021-08-28. Retrieved 2021-05-08.
- ↑ WHO Classification of Tumours Editorial Board, ed. (2020). "13. Mesenchymal tumours of the lower genital tract: Ewing sarcoma". Female genital tumours: WHO Classification of Tumours. Vol. 4 (5th ed.). Lyon (France): International Agency for Research on Cancer. p. 526. ISBN 978-92-832-4504-9. Archived from the original on 2022-06-17. Retrieved 2022-06-23.
- ↑ 11.0 11.1 11.2 Goldman, Lee; Cecil, Russell La Fayette; Schafer, Andrew I (2012). Goldman's Cecil Medicine (24th ed.). Philadelphia: Elsevier Saunders. p. 1326. ISBN 978-1-4377-2788-3. OCLC 909785616. Archived from the original on 2021-08-28. Retrieved 2021-05-08.
- ↑ Asami S, Chin M, Shichino H, Yoshida Y, Nemoto N, Mugishima H, Suzuki T (March 2008). "Treatment of Ewing's sarcoma using an antisense oligodeoxynucleotide to regulate the cell cycle". Biological & Pharmaceutical Bulletin. 31 (3): 391–4. doi:10.1248/bpb.31.391. PMID 18310898.
- ↑ Mateo-Lozano S, Gokhale PC, Soldatenkov VA, Dritschilo A, Tirado OM, Notario V (November 2006). "Combined transcriptional and translational targeting of EWS/FLI-1 in Ewing's sarcoma". Clinical Cancer Research. 12 (22): 6781–90. doi:10.1158/1078-0432.CCR-06-0609. PMID 17121899. S2CID 1404471.
- ↑ Myatt SS, Redfern CP, Burchill SA (April 2005). "p38MAPK-Dependent sensitivity of Ewing's sarcoma family of tumors to fenretinide-induced cell death". Clinical Cancer Research. 11 (8): 3136–48. doi:10.1158/1078-0432.CCR-04-2050. PMID 15837770.
- ↑ Myatt SS, Burchill SA (February 2008). "The sensitivity of the Ewing's sarcoma family of tumours to fenretinide-induced cell death is increased by EWS-Fli1-dependent modulation of p38(MAPK) activity". Oncogene. 27 (7): 985–96. doi:10.1038/sj.onc.1210705. PMID 17700534.
- ↑ Iwamoto Y (February 2007). "Diagnosis and treatment of Ewing's sarcoma". Japanese Journal of Clinical Oncology. 37 (2): 79–89. doi:10.1093/jjco/hyl142. PMID 17272319.
- ↑ Thacker MM, Temple HT, Scully SP (April 2005). "Current treatment for Ewing's sarcoma". Expert Review of Anticancer Therapy. 5 (2): 319–31. doi:10.1586/14737140.5.2.319. PMID 15877528. S2CID 26773908.
- ↑ Bennani-Baiti IM, Cooper A, Lawlor ER, Kauer M, Ban J, Aryee DN, Kovar H (July 2010). "Intercohort gene expression co-analysis reveals chemokine receptors as prognostic indicators in Ewing's sarcoma". Clinical Cancer Research. 16 (14): 3769–78. doi:10.1158/1078-0432.CCR-10-0558. PMC 2905506. PMID 20525755.
- ↑ Sannino G, Marchetto A, Ranft A, Jabar S, Zacherl C, Alba-Rubio R, Stein S, Wehweck FS, Kiran MM, Hoelting TL, Musa J (2018-12-17). "SOX2 expression identifies Ewing sarcoma patients with high risk for tumor relapse and poor survival". bioRxiv: 498253. doi:10.1101/498253. Archived from the original on 2018-12-19. Retrieved 2018-12-18.
- ↑ Twardziok M, Kleinsimon S, Rolff J, Jäger S, Eggert A, Seifert G, Delebinski CI (2016). "Multiple Active Compounds from Viscum album L. Synergistically Converge to Promote Apoptosis in Ewing Sarcoma". PLOS ONE. 11 (9): e0159749. Bibcode:2016PLoSO..1159749T. doi:10.1371/journal.pone.0159749. PMC 5010293. PMID 27589063.
- ↑ "Extraskeletal Ewing sarcoma". www.cancer.gov. 2 February 2011. Archived from the original on 22 January 2022. Retrieved 22 June 2022.
- ↑ Ewing Sarcoma Imaging at eMedicine
- ↑ Veselis, Clinton A.; Awan, Omer; Thomas, Ashanth; Ling, Stephen; Jonnalagadda, Padmaja; Aneja, Amandeep; Ali, Sayed (May 2021). "Bone Tumors Occurring in the Soft Tissues: A Review of the Clinical, Imaging, and Histopathologic Findings". Current Problems in Diagnostic Radiology. 50 (3): 419–429. doi:10.1067/j.cpradiol.2020.06.004. ISSN 1535-6302. PMID 32665061. Archived from the original on 2021-03-30. Retrieved 2022-06-20.
Further reading
- Ewing J (September 2006). "The Classic: Diffuse endothelioma of bone. Proceedings of the New York Pathological Society. 1921;12:17". Clinical Orthopaedics and Related Research. 450: 25–7. doi:10.1097/01.blo.0000229311.36007.c7. PMID 16951641.
External links
| Classification | |
|---|---|
| External resources |
- Pages with script errors
- CS1: long volume value
- All articles with unsourced statements
- Articles with unsourced statements
- Articles with unsourced statements from May 2021
- Articles with invalid date parameter in template
- Articles with unsourced statements from February 2019
- Articles with unsourced statements from January 2013
- Osseous and chondromatous neoplasia
- Small blue round cell tumor
- Sarcoma
- Rare diseases
- RTT