Cryoglobulinemia
| Cryoglobulinemia | |
|---|---|
| Other names: Cryoglobulinaemia, cryoglobulinemic disease | |
 | |
| Specialty | Hematology |
Cryoglobulinemia is a medical condition in which the blood contains large amounts of pathological cold sensitive antibodies called cryoglobulins – proteins (mostly immunoglobulins themselves) that become insoluble at reduced temperatures.[1] This should be contrasted with cold agglutinins, which cause agglutination of red blood cells.
Cryoglobulins typically precipitate (clumps together) at temperatures below normal body temperature – 37 degrees Celsius (99 degrees Fahrenheit) – and will dissolve again if the blood is heated. The precipitated clump can block blood vessels and cause toes and fingers to become gangrenous. While this disease is commonly referred to as cryoglobulinemia in the medical literature, it is better termed cryoglobulinemic disease for two reasons: 1) cryoglobulinemia is also used to indicate the circulation of (usually low levels of) cryoglobulins in the absence of any symptoms or disease and 2) healthy individuals can develop transient asymptomatic cryoglobulinemia following certain infections.[2]
In contrast to these benign instances of circulating cryoglobulins, cryoglobulinemic disease involves the signs and symptoms of precipitating cryoglobulins and is commonly associated with various pre-malignant, malignant, infectious, or autoimmune diseases that are the underlying cause for production of the cryoglobulins.[2][3]
Classification
Since the first description of cryoglobulinemia in association with the clinical triad of skin purpura, joint pain, and weakness by Meltzer et al. in 1967, the percentage of cryoglobulinemic diseases described as essential cryoglobulinemia or idiopathic cryoglobulinemia, that is cryoglobulinemic disease that is unassociated with an underlying disorder, has fallen. Currently most cases of this disease are found to be associated with premalignant, malignant, infectious, or autoimmune disorders that are the known or presumed causes for the production of cryoglobulins. This form of non-essential or non-idiopathic cryoglobulinemic disease is classically grouped into three types according to the Brouet classification.[4] The classification distinguishes three subtypes of cryoglobulinemic diseases based on two factors, the class of immunoglobulins in the cryoglobulin and the association of the cryoglobulinemic disease with other disorder. The following table lists these three types of cryoglobulinemic disease, characterized on the monoclonal immunoglobulin(s) comprising the involved cryoglobulin, percentage of total cryoglobulinemic disease cases, and class of disorders associated for each type.[5][6]
| Type | Composition | Percent of cases | Association with other diseases |
|---|---|---|---|
| Type I | Monoclonal IgG, IgM, IgA, or their κ or λ light chains | 10–15% | Hematological diseases, particularly MGUS, smoldering multiple myeloma, multiple myeloma, Waldenström's macroglobulinemia, and chronic lymphocytic leukemia[7][8] |
| Type II | Monoclonal IgM plus polyclonal IgG or, rarely, IgA | 50–60% | Infectious diseases, particularly hepatitis C infection, HIV infection, and Hepatitis C and HIV coinfection; hematological diseases particularly B cell disorders; autoimmune diseases[7][8] |
| Type III | Polyclonal IgM plus polyclonal IgG or IgA | 25–30% | Autoimmune diseases, particularly Sjögren syndrome and less commonly systemic lupus erythematosus and rheumatoid arthritis; infectious diseases particularly HCV infection[7][8] |
The monoclonal or polyclonal IgM proteins involved in Types II and III cryoglobulinemic disease have rheumatoid factor activity. That is, they bind to polyclonal immunoglobulins, activate the blood complement system, and thereby form tissue deposits that contain IgM, IgG (or, rarely, IgA), and components of the complement system, including in particular complement component 4. The vascular deposition of these types of cryoglobulin-containing immune complexes and complement can cause a clinical syndrome of cutaneous small-vessel vasculitis characterized by systemic vasculitis and inflammation termed cryoglobulinemic vasculitis.[9] Accordingly, type II and type III cryoglobulinemic diseases are often grouped together and referred to as mixed cryoglobulinemia or mixed cryoglobulinemic disease.[8] The monoclonal IgM involved in Type I cryoglobulinemic diseases lacks rheumatoid factor activity.[9]
More recent high resolution protein electrophoresis methods have detected a small monoclonal immunoglobulin component in type III cryoglobulins and/or a micro-heterogeneous composition of oligo-clonal (i.e., more than one monoclonal) immunoglobulin components or immunoglobulins with structures that do not fit into any classifications in the cryoglobulins of ≈10% of type II and III disease cases. It has been proposed that these cases be termed an intermediate type II-III variant of cryoglobulinemic disease and that some of the type III cases associated with the expression of low levels of a one or more isotypes of circulating monoclonal immunoglobulin(s) are in transition to type II disease.[7][10]
Signs and symptoms
The clinical features of cryoglobulinemic disease can reflect those due not only to the circulation of cryoglobulins but also to any underlying hematological premalignant or malignant disorder, infectious disease, or autoimmune syndrome. The following sections of clinical features focuses on those attributed to the cryoglobulins. Cryoglobulins cause tissue damage by three mechanisms; they can:
- a) increase blood viscosity thereby reducing blood flow to tissues to cause the hyperviscosity syndrome (i.e., headache, confusion, blurry or loss of vision, hearing loss, and epistaxis;
- b) deposit in small arteries and capillaries thereby plugging these blood vessels and causing infarction and necrosis of tissues including in particular skin (e.g., ears), distal extremities, and kidneys;
- c) in type II and type III disease, deposit on the epithelium of blood vessels and activate the blood complement system to form pro-inflammatory elements such as C5a thereby initiating the systemic vascular inflammatory reaction termed cryoglobulinemic vasculitis.[2][9]
-
Cryoglobulinemia
-
Cryoglobulinemia
-
Cryoglobulinemia
-
Cryoglobulinemia
-
Cryoglobulinemia
-
Cryoglobulinemia
-
Cryoglobulinemia
-
Cryoglobulinemia
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
Essential cryoglobulinemic disease
The signs and symptoms in the increasingly rare cases of cryoglobulinemic disease that cannot be attributed to an underlying disease generally resemble those of patients suffering Type II and III (i.e., mixed) cryoglobulinemic disease.[9][11]
Type I cryoglobulinemic disease
Signs and symptoms due to the cryoglobulins of type I disease reflect the hyperviscosity and deposition of cryoglobulins within the blood vessels which reduce or stop blood perfusion to tissues. These events occur particularly in cases where blood cryoglobulin levels of monoclonal IgM are high in patients with IgM MGUS, smoldering Waldenström's macroglobulinemia, or Waldenström's macroglobulinemia and in uncommon cases where the levels of monoclonal IgA, IgG, free κ light chains, or free λ light chains are extremely high in patients with non-IgM MGUS, non-IgM smoldering multiple myeloma, or multiple myeloma. The interruption of blood flow to neurological tissues can cause symptoms of confusion, headache, hearing loss, and peripheral neuropathy. Interruption of blood flow to other tissues in type I disease can cause cutaneous manifestations of purpura, blue discoloration of the arms or legs (acrocyanosis), necrosis, ulcers, and livedo reticularis; spontaneous nose bleeds, joint pain, membranoproliferative glomerulonephritis; and cardiovascular disturbances such as shortness of breath, inadequate levels of oxygen in the blood (hypoxemia), and congestive heart failure.[2][9]
Types II and III cryoglobulinemic disease
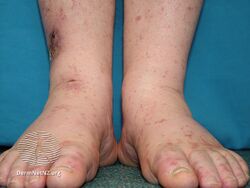
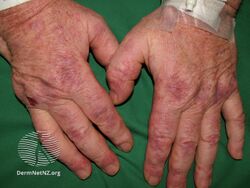
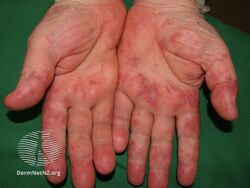
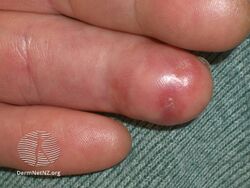
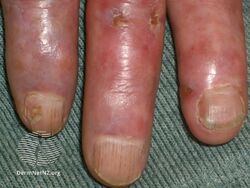
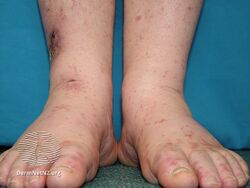
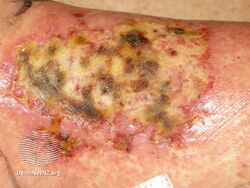
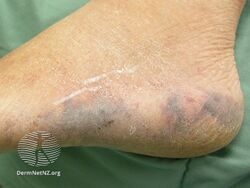
Types II and III (or mixed or variant) cryoglobulinemic disease may also present with symptoms and signs of blood hyperviscosity syndrome and deposition of cryoglobulins within blood vessels but also include those attributable to cryoglobulinemic vasculitis. "Meltzer's triad" of palpable purpura, joint pain, and generalized weakness occurs in ≈33% of patients presenting with type II or type III disease. One or more skin lesions including palpable purpura, ulcers, digital gangrene, and areas of necrosis occur in 69-89% of these mixed disease cases (see attached photograph); less common findings include painful peripheral neuropathy (often manifesting as mononeuritis multiplex in 19-44% of cases), kidney disease (primarily membranoproliferative glomerulonephritis (30%), joint pain (28%), and, less commonly, dry eye syndrome, Raynaud phenomenon (i.e., episodic painful reductions in blood flow to the fingers and toes).[9][12] While the glomerulonephritis occurring in mixed disease appears to be due to inflammatory vasculitis, the glomerulonephritis occurring in type I disease appears due to the interruption of blood flow.[12] The hematological, infectious, and autoimmune diseases underlying type II cryoglobulinemic disease and the infectious and autoimmune diseases underlying type III cryoglobulinemic disease are also critical parts of the disease's clinical findings.
Mechanism
Cryoglobulins
Cryoglobulins consists of one or more of the following components: monoclonal or polyclonal IgM, IgG, IgA antibodies, monoclonal κ, or λ free light chain portions of these antibodies, and proteins of the blood complement system, particularly complement component 4 (C4). The particular components involved are a reflection of the disorders which are associated with, and considered to be the cause of, the cryoglobulinemic disease. The cryoglobulin compositions and disorder associations in cryoglobulinemic disease are as follows:
- Monoclonal IgM-based cryogloblin occurs in cases of Waldenström's macroglobulinemia and the pre-malignant precursors to this cancer, IgM monoclonal gammopathy of undetermined significance and smoldering Waldenstrom's macroglobulinemia.[2]
- Monoclonal IgG or, rarely, IgA, κ light chain, or λ light chain cryoglobulins occur in cases of multiple myeloma and the pre-malignant precursors to this cancer, non-IgM monoclonal gammopathy of undetermined significance and non-IgM smoldering multiple myeloma. Non-IgM monoclonal immunoglobulin-based cases of cryoglobulinemic disease are less commonly associated with other B-cell lymphocytic diseases viz., Non-Hodgkin lymphoma, Hodgkin lymphoma, B-cell chronic lymphocytic leukemia, and Castleman disease; they occur rarely in non-B cell hematological disorders such as myelodysplastic syndromes and chronic myelogenous leukemia.[7] Among these purely monoclonal immunoglobulin causes of cryoglobulinemic disease, Waldenström macroglobulinemia and multiple myeloma together account for ≈40% of cases; their pre-malignant precursors account for ≈44% of cases; and the other cited hematological diseases account for ≈16% of cases.[2][3]
- Mixtures of monoclonal or polyclonal IgM, IgG, and/or IgA along with blood complement proteins such as C4 are the cryoglobulins associated with cases of infectious diseases, particularly hepatitis C infection, HIV infection, and Hepatitis C and HIV coinfection, and, less commonly or rarely, with cases of other infectious diseases such as hepatitis B infection, hepatitis A infection, cytomegalovirus infection, Epstein–Barr virus infection, Lyme disease, syphilis, lepromatous leprosy, Q fever, poststreptococcal nephritis, subacute bacterial endocarditis, coccidioidomycosis, malaria, schistosomiasis, echinococcosis, toxoplasmosis, and Kala-azar. These mixed-protein cryoglobulins are also associated with autoimmune diseases, particularly Sjögren syndrome, less commonly systemic lupus erythematosus and rheumatoid arthritis, and rarely polyarteritis nodosa, systemic sclerosis, temporal arteritis, polymyositis, Henoch–Schönlein purpura, pemphigus vulgaris, sarcoidosis, inflammatory bowel diseases, and others.[7] In these mixed-protein depositions, the monoclonal or polyclonal IgM typically possesses rheumatoid factor activity and therefore binds to the Fc region of polyclonal IgG antibodies, activates the blood complement system, and complexes with complement components to form precipitates composed of IgM, IgG or IgG, and complement components, particularly complement component 4 (C4).[3]
Diagnosis
Cryoglobulinemia and cryoglobulinemic disease must be distinguished from cryofibrinogenemia or cryofibrinogenemic disease, conditions which involve the cold-induced intravascular deposition of circulating native fibrinogens.[13][14] These molecules precipitate at lower temperatures (e.g., 4 °C). Since cryofibrinogens are present in plasma but greatly depleted in serum, precipitation tests for them are positive in plasma but negative in serum.[14] Cryofibrinogenemia is occasionally found in cases of cryoglobulinemic disease.[15] Cryoglobulinemic disease must also be distinguished from frostbite as well as numerous other conditions that have a clinical (particularly cutaneous) presentation similar to cryoglobulinemic disease but are not exacerbated by cold temperature, e.g., dysfibrinogenemia and dysfibrinogenemic disease (conditions involving the intravascular deposition of genetically abnormal circulating fibrinogens), purpura fulminans, cholesterol emboli, warfarin necrosis, ecthyma gangrenosum, and various hypercoagulable states.[15]
Rheumatoid factor is a sensitive test for cryoglobulinemia. The precipitated cryoglobulins are examined by immunoelectrophoresis and immunofixation to detect and quantify the presence of monoclonal IgG, IgM, IgA, κ light chain, or λ light chain immunoglobins. Other routine tests include measuring blood levels of rheumatoid factor activity, complement C4, other complement components, and hepatitic C antigen. Biopsies of skin lesions and, where indicated, kidney or other tissues can help in determining the nature of the vascular disease (immunoglobulin deposition, cryoglobulinemic vasculitis, or, in cases showing the presence of cryfibrinogenemia, fibrinogen deposition. In all events, further studies to determine the presence of hematological, infections, and autoimmune disorders are conducted on the basis of these findings as well as each cases clinical findings.[2][12][15]
Treatment
All patients with symptomatic cryoglobulinemia are advised to avoid, or protect their extremities, from exposure to cold temperatures. Refrigerators, freezers, and air-conditioning represent dangers of such exposure.[12][13]
Asymptomatic cryoglobulinemia
Individuals found to have circulating cryoglobulins but no signs or symptoms of cryoglobulinemic diseases should be evaluated for the possibility that their cryoglobulinemia is a transient response to a recent or resolving infection. Those with a history of recent infection that also have a spontaneous and full resolution of their cryoglobulinemia need no further treatment. Individuals without a history of infection and not showing resolution of their cryoglobulinemia need to be further evaluated. Their cryoglobulins should be analyzed for their composition of immunoglobulin type(s) and complement component(s) and examined for the presence of the premalignant and malignant diseases associated with Type I disease as well as the infectious and autoimmune diseases associated with type II and type III disease.[12] A study conducted in Italy on >140 asymptomatic individuals found five cases of hepatitis C-related and one case of hepatitis b-related cryoglobulinemia indicating that a complete clinical examination of asymptomatic individuals with cryoglobulinemia offers a means for finding people with serious but potentially treatable and even curable diseases.[16] Individuals who show no evidence of a disease underlying their cryoglobulinemia and who remain asymptomatic should be followed closely for any changes that may indicate development of cryoglobulinemic disease.[16]
Severely symptomatic cryoglobulinemic disease
People affected by the severest, often life-threatening, complications of cryoglobulinemic disease require urgent plasmapharesis and/or plasma exchange in order to rapidly reduce the circulating levels of their cryoglobulins. Complications commonly requiring this intervention include: hyperviscosity disease with severe symptoms of neurological (e.g., stroke, mental impairment, and myelitis) and/or cardiovascular (e.g., congestive heart failure, myocardial infarction) disturbances; vasculitis-driven intestinal ischemia, intestinal perforation, cholecystitis, or pancreatitis, causing acute abdominal pain, general malaise, fever, and/or bloody bowel movements; vasculitis-driven pulmonary disturbances (e.g., coughing up blood, acute respiratory failure, X-ray evidence of diffuse pulmonary infiltrates caused by diffuse alveolar hemorrhage); and severe kidney dysfunction due to intravascular deposition of immunoglobulins or vasculitis. Along with this urgent treatment, severely symptomatic patients are commonly started on therapy to treat any underlying disease; this treatment is often supplemented with anti-inflammatory drugs such as corticosteroids (e.g., dexamethasone) and/or immunosuppressive drugs. Cases where no underlying disease is known are also often treated with the latter corticosteroid and immunosuppressive medications.[2][7][13]
Type I cryoglobulinemic disease
Treatment of Type I disease is generally directed towards treating the underlying pre-malignant or malignant disorder (see plasma cell dyscrasia, Waldenström's macroglobulinemia, and chronic lymphocytic leukemia). This involves appropriate chemotherapy regimens which may include bortezomib (promotes cell death by apoptosis in cells accumulating immunoglobulins) in patients with monoclonal immunoglobulin-induced kidney failure and rituximab (antibody directed against CD20 surface antigen-bearing lymphocytes) in patients with Waldenstroms macroglobulonemia).[12][13]
Type II and III cryoglobulinemic disease
Treatment of mixed cryoglobulinemic disease is, similar to type I disease, directed toward treating any underlying disorder. This includes malignant (particularly Waldenström's macroglobulinemia in type II disease), infectious, or autoimmune diseases in type II and III disease. Recently, evidence of hepatitis C infection has been reported in the majority of mixed disease cases with rates being 70-90% in areas with high incidences of hepatitis C.[9] The most effective therapy for hepatitis C-associated cryoglobulinemic disease consists of a combination of anti-viral drugs, pegylated INFα and ribavirin; depletion of B cells using rituximab in combination with antiviral therapy or used alone in patients refractory to antiviral therapy has also proven successful in treating the hepatitis C-associated disease.[2][12] Data on the treatment of infectious causes other than hepatitis C for the mixed disease are limited. A current recommendation treats the underlying disease with appropriate antiviral, anti-bacterial, or anti-fungal agents, if available; in cases refractory to an appropriate drug, the addition of immunosuppressive drugs to the therapeutic regimen may improve results.[12] Mixed cryoglobulinemic disease associated with autoimmune disorders is treated with immunosuppressive drugs: combination of a corticosteroid with either cyclophosphamide, azathioprine, or mycophenolate or combination of a corticosteroid with rituximab have been used successfully to treated mixed disease associated with autoimmune disorders.[2][12]
See also
- Cryofibrinogenemia
- Cryoglobulinemic purpura
- Cryoglobulinemic vasculitis
- Dysfibrinogenemia
- Hematopoietic ulcer
- Hyperviscosity syndrome
- Paraproteinemia
- Plasma cell dyscrasias
References
- ↑ "Cryoglobulinemia" at Dorland's Medical Dictionary
- ↑ 2.0 2.1 2.2 2.3 2.4 2.5 2.6 2.7 2.8 2.9 Retamozo S, Brito-Zerón P, Bosch X, Stone JH, Ramos-Casals M (2013). "Cryoglobulinemic disease". Oncology (Williston Park, N.Y.). 27 (11): 1098–1105, 1110–6. PMID 24575538.
- ↑ 3.0 3.1 3.2 Ghetie D, Mehraban N, Sibley CH (2015). "Cold hard facts of cryoglobulinemia: updates on clinical features and treatment advances". Rheumatic Diseases Clinics of North America. 41 (1): 93–108, viii–ix. doi:10.1016/j.rdc.2014.09.008. PMID 25399942.
- ↑ Brouet JC, Clauvel JP, Danon F, Klein M, Seligmann M (1974). "Biologic and clinical significance of cryoglobulins. A report of 86 cases". Am. J. Med. 57 (5): 775–88. doi:10.1016/0002-9343(74)90852-3. PMID 4216269.
- ↑ Ferri C, Zignego AL, Pileri SA (2002). "Cryoglobulins". J. Clin. Pathol. 55 (1): 4–13. doi:10.1136/jcp.55.1.4. PMC 1769573. PMID 11825916.
- ↑ "Cryoglobulin" at Dorland's Medical Dictionary
- ↑ 7.0 7.1 7.2 7.3 7.4 7.5 7.6 Tedeschi A, Baratè C, Minola E, Morra E (2007). "Cryoglobulinemia". Blood Reviews. 21 (4): 183–200. doi:10.1016/j.blre.2006.12.002. PMID 17289231.
- ↑ 8.0 8.1 8.2 8.3 Tedeschi A, Baratè C, Minola E, Morra E (2007). "Cryoglobulinemia". Blood Rev. 21 (4): 183–200. doi:10.1016/j.blre.2006.12.002. PMID 17289231.
- ↑ 9.0 9.1 9.2 9.3 9.4 9.5 9.6 Ostojic P, Jeremic IR (2017). "Managing refractory cryoglobulinemic vasculitis: challenges and solutions". Journal of Inflammation Research. 10: 49–54. doi:10.2147/JIR.S114067. PMC 5428757. PMID 28507447.
- ↑ Tissot JD, Schifferli JA, Hochstrasser DF, et al. (1994). "Two-dimensional polyacrylamide gel electrophoresis analysis of cryoglobulins and identification of an IgM-associated peptide". J. Immunol. Methods. 173 (1): 63–75. doi:10.1016/0022-1759(94)90284-4. PMID 8034987.
- ↑ "Overview of cryoglobulins and cryoglobulinemia". www.uptodate.com. Archived from the original on August 2, 2017. Retrieved August 31, 2017.
- ↑ 12.0 12.1 12.2 12.3 12.4 12.5 12.6 12.7 12.8 Muchtar E, Magen H, Gertz MA (2017). "How I treat cryoglobulinemia". Blood. 129 (3): 289–298. doi:10.1182/blood-2016-09-719773. PMID 27799164.
- ↑ 13.0 13.1 13.2 13.3 Michaud M, Pourrat J (2013). "Cryofibrinogenemia". Journal of Clinical Rheumatology. 19 (3): 142–8. doi:10.1097/RHU.0b013e318289e06e. PMID 23519183.
- ↑ 14.0 14.1 Caimi G, Carlisi M, Urso C, Lo Presti R, Hopps E (2017). "Clinical disorders responsible for plasma hyperviscosity and skin complications". European Journal of Internal Medicine. 42: 24–28. doi:10.1016/j.ejim.2017.04.001. PMID 28390781.
- ↑ 15.0 15.1 15.2 Grada A, Falanga V (2017). "Cryofibrinogenemia-Induced Cutaneous Ulcers: A Review and Diagnostic Criteria". American Journal of Clinical Dermatology. 18 (1): 97–104. doi:10.1007/s40257-016-0228-y. PMID 27734332. S2CID 39645385.
- ↑ 16.0 16.1 Monti G, Saccardo F, Castelnovo L, Novati P, Sollima S, Riva A, Sarzi-Puttini P, Quartuccio L, De Vita S, Galli M (2014). "Prevalence of mixed cryoglobulinaemia syndrome and circulating cryoglobulins in a population-based survey: the Origgio study". Autoimmunity Reviews. 13 (6): 609–14. doi:10.1016/j.autrev.2013.11.005. PMID 24418294.
External links
| Classification | |
|---|---|
| External resources |