Chondroblastoma
| Chondroblastoma | |
|---|---|
 | |
| Chondoblastoma of thigh bone near knee (CT scan, non-contrast) | |
| Symptoms | Pain, joint swelling, reduced movement, muscle wasting[1] |
| Usual onset | Children and young adults (<20)[1] |
| Risk factors | Unknown[1] |
| Diagnostic method | X-ray, computed tomography (CT), magnetic resonance imaging (MRI),[1] bone scans, biopsy |
| Treatment | surgical curettage[2] |
| Prognosis | Good with treatment,[2] 5-12% recur[2] |
| Frequency | Rare, <1% of all bone tumors, 2M:F[1] |
Chondroblastoma is a type of non-cancerous bone tumor of the chondrogenic type.[3] It typically affects the ends of long bones or their apophyses.[1][4] It generally presents with pain, sometimes present for years, which begins mild and progressively become severe.[5] There may be joint swelling.[5]
It is thought to arise from an outgrowth of immature cartilage cells (chondroblasts) from secondary ossification centers, originating from the epiphyseal plate or some remnant of it.[6][7]
Diagnosis involves medical imaging.[8] It can be better distinguished using CT scan or MRI.[9] Treatment is with surgical curettage.[10] Most are cured but 5-12% may recur.[2]
Chondroblastoma accounts for around 1% of all bone tumors.[11] It affects mostly children and young adults in their 20s or younger.[10][12] It affects twice as many males than females.[1] The most commonly affected sites are the femur, humerus and tibia.[11] Less commonly affected sites include the talus and calcaneus of the foot and flat bones.[10][13] Chondroblastoma was first described in 1927.[10][12]
Signs and Symptoms
_(Radiopaedia_9270).png)
The most common symptom is pain, which may be initially attributed to a minor injury or sports-related injury, start off mild and progressively become severe.[5] If the chondroblastoma is near a joint, the joint may become swollen and difficult to move.[5] Pain may be present for several weeks, months, or years.[6][10] Other symptoms in order of most common to least commonly observed include swelling, a limp (when affected bone is in the lower extremity), joint stiffness, and a lump.[10][13]
Physical findings include localized tenderness and a decreased range of motion in the involved bone and nearby joint, muscle atrophy, a palpable mass, soft tissue swelling, and joint effusion in the affected area.[10][13][14] Less commonly, pathological fractures can be found, especially in cases involving the foot.[10] In cases involving the temporal bone, tinnitus, dizziness, and hearing loss have been reported.[13]
In a publication by Turcotte et al. it was found that the average duration of symptoms for people with chondroblastoma was about 20 months, ranging from 5 weeks to 16 years.[5][13]
Causes
Currently, the genetic or environmental factors that predispose an individual for chondroblastoma are not well known or understood.[10] Furthermore, it is most often observed in young patients that are skeletally immature, with most cases diagnosed in the second decade of life.[6][14] Approximately 92% of patients presenting with chondroblastoma are younger than 30 years.[14] There is no indication of a racial predilection for chondroblastoma.[14]
Mechanism
The etiology of chondroblastoma is uncertain, as there is no specific characteristic abnormality or chromosomal breaking point observed, despite cytogenetic abnormalities being highly specific for some tumors.[10][14]
Romeo et al has noted that chondroblastoma arising in long bones mainly affects the epiphyses, while in other locations it is close to ossification centers.[6] Additionally, rare prevalence of chondroblastoma in intra-membranous ossification suggests a close relationship with growth plate cartilage.[6] In chondroblastoma, growth signaling molecules may be present due to the pre-pubertal signaling network as well as cartilage growth.[6] Sex hormones are thought to be linked to this process because of the spatial relationship of chondroblastoma with the growth plate and its typical occurrence before growth plate fusion.[6] Both Indian Hedgehog/Parathyroid Hormone-related Protein (IHh/PtHrP) and fibroblast growth factor (FGF) signaling pathways, important for development of the epiphyseal growth plate, are active in chondroblastoma leading to greater proliferation among the cells in the proliferating/pre-hypertrophic zone (cellular-rich area) versus the hypertrophic/calcifying zone (matrix-rich area).[10][6] These findings suggest that chondroblastoma is derived from a mesenchymal cell undergoing chondrogenesis via active growth-plate signaling pathways (see Endochondral ossification).[10][6]
The highly heterogeneous nature of the tumor makes classification particularly difficult especially considering the origins of chondroblastoma.[6] There are two opposing views on the nature of chondroblastoma, one favoring an osseous origin and the other favoring a cartilaginous origin. The work of Aigner et al suggests that chondroblastoma should be reclassified as a bone-forming neoplasm versus a cartilaginous neoplasm due to the presence of osteoid matrix, type I collagen, and absence of true cartilage matrix (collagen II).[6][14] However, Edel et al found that collagen II, a marker for mature chondrocytes, was expressed in chondroblastoma, supporting the chondroid nature of the neoplasm.[6] The results of Romeo and colleagues favor the view of Edel et al of chondroblastoma being cartilaginous in nature but recognize that any definitive determinations regarding the origin of this neoplasm are not possible because of the plasticity of mesenchymal cells when set into different microenvironments and static approaches used in literature.[6] Romeo et al have observed chondroblastoma neoplasms to be composed of mesenchymal cells that have completed normal chondrogenesis along with the production of osteoid and collagen I that could be the result of transdifferentiation of chondrocytes towards osteoblasts.[6]
Diagnosis
Imaging Studies
A variety of imaging studies can be used to diagnose chondroblastoma, with radiographs being the most common.[10][8] Laboratory studies are not considered useful.[14] Classical chondroblastoma (appearing on long bones) appears as a well-defined eccentric oval or round lytic lesion that usually involves the adjacent bone cortex without periosteal reaction.[10][13] A sclerotic margin can be seen in some cases.[10][13] For long bone chondroblastomas the tumor is typically contained to the epiphysis or apophysis but may extend through the epiphyseal plate.[10][13] Chondroblastomas are usually located in the medullary portion of bones and can, in some cases, include the metaphysis.[10][13] However, true metaphyseal chondroblastomas are rare and are typically the result of an extension from a neighboring epiphyseal legion.[10][13] Most lesions are less than 4 cm.[10] A mottled appearance on the radiograph is not atypical and indicates areas of calcification which is commonly associated with skeletally immature patients.[10] Additionally, one-third of all cases involve aneurysmal bone cysts which are thought to be the result of stress, trauma or hemorrhage.[10] In cases involving older patients or flat bones, typical radiographic presentation is not as common and may mimic aggressive processes.[10][13]
Other imaging techniques involve computed tomography (CT), magnetic resonance imaging (MRI), and bone scans, which may be helpful in determining the anatomical boundaries, associated edema, or biological activity of the chondroblastoma, respectively.[8][10] MRI studies may show extensive oedema around the lesion and show variable T2 signal intensity.[15]
-
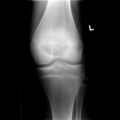
1. a. X-ray of chondroblastoma of thigh bone near knee
-
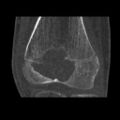
1. b. CT scan shows chondroblastoma of thigh bone near knee more clearly
-
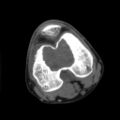
1. c. CT scan of chondroblastoma of thigh bone near knee (cross-section view)
-
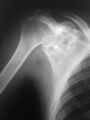
2. X-ray of chondroblastoma of shoulder blade
-
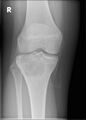
3. X-ray of chondroblastoma of large long bone of lower leg, near the knee



.jpg)

Histological Findings

Chondroid differentiation is a common feature of chondroblastoma.[10][12][13] A typical histological appearance consists of a combination of oval mononuclear and multi-nucleated osteoclast-type giant cells.[10][7][12] However this is not a prerequisite for diagnosis, as cells with epithelioid characteristics have been observed in lesions of the skull and facial bones.[12] A "chicken-wire" appearance is characteristic of chondroblastoma cells and is the result of dystrophic calcification that may surround individual cells.[10][14] Although, calcification may not be present and is not a prerequisite for diagnosis.[10][7][12] Mitotic figures can be observed in chondroblastoma tissue but are not considered atypical in nature, and therefore, should not be viewed as a sign of a more serious pathology.[10][12] There is no correlation between mitotic activity and location of the lesion.[12] Furthermore, the presence of atypical cells is rare and is not associated with malignant chondroblastoma.[10][13] There are no discernible histological differences observed when comparing the aggressive form of chondroblastoma that can cause recurrence or metastases with its less aggressive, benign, counterpart.[citation needed]
Differential Diagnosis
Chondromyxoid fibromas can share characteristics with chondroblastomas with regards to histologic and radiographic findings. However they more commonly originate from the metaphysis, lack calcification and have a different histologic organization pattern.[14] Other differential diagnoses for chondroblastoma consist of giant cell tumors, bone cysts, eosinophilic granulomas, clear cell chondrosarcomas, and enchondromas (this list is not exhaustive).[10][14]
Treatment
Chondroblastoma has not been known to spontaneously heal and the standard treatment is surgical curettage of the lesion with bone grafting.[10] To prevent recurrence or complications it is important to excise the entire tumor following strict oncologic criteria.[10][16] However, in skeletally immature patients intraoperative fluoroscopy may be helpful to avoid destruction of the epiphyseal plate.[10] In patients who are near the end of skeletal growth, complete curettage of the growth plate is an option.[10] In addition to curettage, electric or chemical cauterization (via phenol) can be used as well as cryotherapy and wide or marginal resection.[10][16] Depending on the size of the subsequent defect, autograft or allograft bone grafts are the preferred filling materials.[10][16] Other options include substituting polymethylmethacrylate (PMMA) or fat implantation in place of the bone graft.[10][7][16] The work of Ramappa et al suggests that packing with PMMA may be a more optimal choice because the heat of polymerization of the cement is thought to kill any remaining lesion.[10][7]
Both radiotherapy and chemotherapy are not commonly used.[10][16] Radiotherapy has been implemented in chondroblastoma cases that are at increased risk of being more aggressive and are suspected of malignant transformation.[10][16] Furthermore, radiofrequency ablation has been used, but is typically most successful for small chondroblastoma lesions (approximately 1.5 cm).[10] Treatment with radiofrequency ablation is highly dependent on size and location due to the increased risk of larger, weight-bearing lesions being at an increased risk for articular collapse and recurrence.[10][16]
Overall, the success and method of treatment is highly dependent upon the location and size of the chondroblastoma.[10][12][16]
Outcomes
Although not specific to one mode of management, lesion size, patient sex, or follow-up, the recurrence rate for chondroblastoma is relatively high, and has been shown in select studies to be dependent upon the anatomical location, method of treatment, and biological aggressiveness of the initial lesion.[10][7][14] The rate of recurrence is highly variable, ranging between 5% and 40%, as study results are generally inconclusive.[10] However, local recurrence for long bone lesions is around 10%, with chondroblastoma in flat bones having higher recurrence and more complications.[10][14] Recurrences are more common in cases involving an open epiphyseal plate where they can be attributed to inadequate curettage to avoid damage.[10][14] Lesions of the proximal femur are particularly problematic because of difficulties accessing the femoral head for complete excision.[10] Chondroblastoma may recur in the soft tissue surrounding the initial lesion, especially in the case of incomplete curettage.[10] Recurrences have been shown to occur between 5 months and 7 years after initial treatment and are generally treated with repeat curettage and excision of affected soft-tissue.[10][14] No histological differences have been seen between recurrent and non-recurrent chondroblastomas.[10][12][13]
Rarely, more aggressive chondroblastomas can metastasize.[10] The most common location for metastases is the lung, with some cases also involving secondary bone sites, soft tissue, skin, or the liver.[10][14] The prevalence of metastatic chondroblastoma, however, is quite low and is believed to be less than 1%.[10] There is no relationship established between metastasis and previous surgery, non-surgical treatment, anatomical location, or patient age.[10] Survival of patients with metastatic lesions is better when the metastases are surgically resectable, as chemotherapy has been shown to have little to no benefit.[10] Prognosis is bleak for patients with malignant chondroblastomas that are resistant to surgery, radiation, and chemotherapy.[14] However, patients with resectable metastases have survived for several years following diagnosis.[10]
While recurrence is the most common complication of chondroblastoma other issues include post-surgery infection, degenerative joint disease, pathological fractures, failure of bone grafts, pre-mature epiphyseal closure, functional impairment, and malignant transformation.[10][14] Complications are less common in patients presenting with chondroblastoma in accessible areas.[10] Overall, patients with more classical chondroblastoma (appearing in long bones, typical presentation) have better prognoses than patients with atypical chondroblastoma (flat bones, skull, etc.).[10][7][12][14][13]
Epidemiology
Chondroblastoma comprise less than 1% of all bone tumors,[1] and around 5% of cartilage tumors.[17] It affects twice as many males than females.[1]
History
Chondroblastoma was first described in 1927 as a cartilage-containing giant cell tumor by Kolodny but later characterized by Codman in 1931.[10][12] Codman believed chondroblastoma to be an "epiphyseal chondromatous giant cell tumor" in the proximal humerus.[10][7] This view was changed later by a comprehensive review completed by Jaffe and Lichtenstein in 1942 of similar tumors in other locations than the proximal humerus.[10][12] They re-defined the tumor as a benign chondroblastoma of the bone that is separate from giant cell tumors.[11] However, chondroblastoma of the proximal humerus is still sometimes referred to as Codman’s Tumor.[10][7][12]
References
- ↑ 1.0 1.1 1.2 1.3 1.4 1.5 1.6 1.7 1.8 WHO Classification of Tumours Editorial Board, ed. (2020). "Chondroblastoma". Soft Tissue and Bone Tumours: WHO Classification of Tumours. Vol. 3 (5th ed.). Lyon (France): International Agency for Research on Cancer. pp. 359–361. ISBN 978-92-832-4503-2. Archived from the original on 2021-06-13. Retrieved 2021-05-10.
- ↑ 2.0 2.1 2.2 2.3 Suster, David; Hung, Yin Pun; Nielsen, G. Petur (1 January 2020). "Differential Diagnosis of Cartilaginous Lesions of Bone". Archives of Pathology & Laboratory Medicine. 144 (1): 71–82. doi:10.5858/arpa.2019-0441-RA. ISSN 0003-9985. Archived from the original on 19 July 2021. Retrieved 17 July 2021.
- ↑ WHO Classification of Tumours Editorial Board, ed. (2020). "Bone tumors". Soft Tissue and Bone Tumours: WHO Classification of Tumours. Vol. 3 (5th ed.). Lyon (France): International Agency for Research on Cancer. p. 338. ISBN 978-92-832-4503-2. Archived from the original on 2021-06-13. Retrieved 2021-05-10.
- ↑ "ICD-11 - ICD-11 for Mortality and Morbidity Statistics". icd.who.int. Archived from the original on 1 August 2018. Retrieved 24 June 2021.
- ↑ 5.0 5.1 5.2 5.3 5.4 Damron, Timothy A. (25 September 2020). "Chondroblastoma Clinical Presentation: History, Physical Examination". emedicine.medscape.com. Archived from the original on 4 May 2021. Retrieved 4 May 2021.
- ↑ 6.00 6.01 6.02 6.03 6.04 6.05 6.06 6.07 6.08 6.09 6.10 6.11 6.12 6.13 Romeo, S.; Bovée, Jvmg; Jadnanansing, N. a. A.; Taminiau, A. H. M.; Hogendoorn, P. C. W. (2004). "Expression of cartilage growth plate signalling molecules in chondroblastoma". The Journal of Pathology. 202 (1): 113–120. doi:10.1002/path.1501. ISSN 1096-9896. Archived from the original on 2021-08-28. Retrieved 2021-05-04.
- ↑ 7.0 7.1 7.2 7.3 7.4 7.5 7.6 7.7 7.8 Ramappa, Arun; Lee, Ncis Y.; Tang, Peter (September 2000). "Chondroblastoma of Bone". The Journal of Bone and Joint Surgery. 82-A (8): 1140–5. doi:10.2106/00004623-200008000-00011. Archived from the original on 2021-08-28. Retrieved 2021-05-04.
- ↑ 8.0 8.1 8.2 Damron, Timothy A. (25 September 2020). "Chondroblastoma workup". emedicine.medscape.com. Archived from the original on 4 May 2021. Retrieved 4 May 2021.
- ↑ Bocklage, Therese J.; Quinn, Robert; Verschraegen, Claire; Schmit, Berndt (2014). "16. Cartilaginous tumours of bones and joints". Bone and Soft Tissue Tumors: A Multidisciplinary Review with Case Presentations. London: JP Medical Ltd. pp. 366–409. ISBN 978-1-907816-22-2. Archived from the original on 2021-07-09. Retrieved 2021-08-21.
- ↑ 10.00 10.01 10.02 10.03 10.04 10.05 10.06 10.07 10.08 10.09 10.10 10.11 10.12 10.13 10.14 10.15 10.16 10.17 10.18 10.19 10.20 10.21 10.22 10.23 10.24 10.25 10.26 10.27 10.28 10.29 10.30 10.31 10.32 10.33 10.34 10.35 10.36 10.37 10.38 10.39 10.40 10.41 10.42 10.43 10.44 10.45 10.46 10.47 10.48 10.49 10.50 10.51 10.52 10.53 10.54 10.55 10.56 10.57 10.58 10.59 10.60 10.61 10.62 10.63 De Mattos, Camila B. R.; Angsanuntsukh, Chanika; Arkader, Alexandre; Dormans, John P. (April 2013). "Chondroblastoma and chondromyxoid fibroma". The Journal of the American Academy of Orthopaedic Surgeons. 21 (4): 225–233. doi:10.5435/JAAOS-21-04-225. ISSN 1067-151X. PMID 23545728. Archived from the original on 2021-08-28. Retrieved 2021-05-04.
- ↑ 11.0 11.1 11.2 Damron, Timothy A. (25 September 2020). "Chondroblastoma: Background, Pathophysiology, Etiology". emedicine.medscape.com. Archived from the original on 4 May 2021. Retrieved 4 May 2021.
- ↑ 12.00 12.01 12.02 12.03 12.04 12.05 12.06 12.07 12.08 12.09 12.10 12.11 12.12 12.13 Kurt, Ann-Marie, et al. "Chondroblastoma of Bone." Human Pathology 20.10 (1989): 965-976. Web. 5 Dec. 2015.
- ↑ 13.00 13.01 13.02 13.03 13.04 13.05 13.06 13.07 13.08 13.09 13.10 13.11 13.12 13.13 13.14 Turcotte, Robert E., et al. "Chondroblastoma." Human Pathology 24.9 (1993): 944-949. Web. 6 Dec. 2015.
- ↑ 14.00 14.01 14.02 14.03 14.04 14.05 14.06 14.07 14.08 14.09 14.10 14.11 14.12 14.13 14.14 14.15 14.16 14.17 Damron, Timothy A. "Chondroblastoma." MedScape (2014). Web. 6 Dec. 2015
- ↑ Chen, Wenqian; DiFrancesco, Lisa M. (June 2017). "Chondroblastoma: An Update". Archives of Pathology & Laboratory Medicine. 141 (6): 867–871. doi:10.5858/arpa.2016-0281-RS. ISSN 0003-9985. Archived from the original on 2021-08-28. Retrieved 2020-07-23.
- ↑ 16.0 16.1 16.2 16.3 16.4 16.5 16.6 16.7 Damron, Timothy A. (25 September 2020). "Chondroblastoma Treatment & Management". emedicine.medscape.com. Archived from the original on 4 May 2021. Retrieved 4 May 2021.
- ↑ Engel, Hannes; Herget, Georg W.; Füllgraf, Hannah; Sutter, Reto; Benndorf, Matthias; Bamberg, Fabian; Jungmann, Pia M. (March 2021). "Chondrogenic Bone Tumors: The Importance of Imaging Characteristics". RoFo: Fortschritte Auf Dem Gebiete Der Rontgenstrahlen Und Der Nuklearmedizin. 193 (3): 262–275. doi:10.1055/a-1288-1209. ISSN 1438-9010. PMID 33152784. Archived from the original on 2021-07-09. Retrieved 2021-07-05.
External links
| Classification | |
|---|---|
| External resources |