Carnitine-acylcarnitine translocase deficiency
| Carnitine-acylcarnitine translocase deficiency, Carnroviatonis | |
|---|---|
| Other names: CATD, Carnroviatonis | |
 | |
| Staining of liver tissue shows extensive vacuolar degeneration | |
Carnitine-acylcarnitine translocase deficiency is a rare, autosomal recessive metabolic disorder that prevents the body from converting long-chain fatty acids into energy, particularly during periods without food.[1] Carnitine, a natural substance acquired mostly through the diet, is used by cells to process fats and produce energy. People with this disorder have a faulty enzyme that prevents long-chain fatty acids from being transported into the innermost part of the mitochondria for processing.[citation needed]
Signs and symptoms
The signs of carnitine-acylcarnitine translocase deficiency usually begin within the first few hours of life. Seizures, an irregular heartbeat, and breathing problems are often the first signs of this disorder. This disorder may also cause extremely low levels of ketones (products of fat breakdown that are used for energy) and low blood sugar (hypoglycemia). Together, these two signs are called hypoketotic hypoglycemia. Other signs that are often present include ammonia in the blood (hyperammonemia), an enlarged liver (hepatomegaly), heart abnormalities (cardiomyopathy), and muscle weakness. This disorder can cause sudden infant death.[citation needed]
Pathophysiology
Mutations in the SLC25A20 gene lead to the production of a defective version of an enzyme called carnitine-acylcarnitine translocase.
Without this enzyme, long-chain fatty acids from food and fats stored in the body cannot be broken down and processed. As a result, these fatty acids are not converted into energy, which can lead to characteristic signs and symptoms of this disorder, such as weakness, hypoglycemia, and an irregular heartbeat. Free long-chain fatty acids or those that are joined with carnitine can affect the electrical properties of cardiac cells causing an irregular heart beat (arrhythmia, which can lead to cardiac arrest). Fatty acids may also build up in tissues and can damage the heart, liver, and muscles, and cause more serious complications.[citation needed]
This condition has an autosomal recessive inheritance pattern, which means the defective gene is located on an autosome, and two copies of the gene - one from each parent - must be inherited to be affected by the disorder. The parents of a child with an autosomal recessive disorder are carriers of one copy of the defective gene, but are usually not affected by the disorder.[citation needed]
-

Acyl-CoA from cytosol to the mitochondrial matrix
-
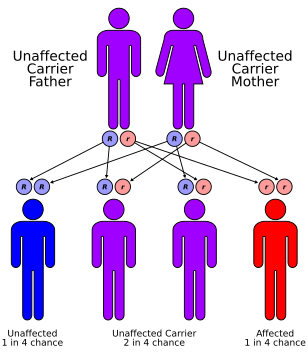
Carnitine-acylcarnitine translocase deficiency has an autosomal recessive pattern of inheritance.


Diagnosis
In terms of the diagnosis of Carnitine-acylcarnitine translocase deficiency the following is done:[2]
- Clinical findings
- Genetic test
Differential diagnosis
The DDx is based on the following:[2]
- Very long-chain acyl-CoA dehydrogenase deficiency
- Carnitine palmitoyltransferase II deficiency
- TANGO2-related metabolic encephalopathy and arrhythmias
Treatment
In terms of management of Carnitine-acylcarnitine translocase deficiency one needs to avoid fasting. Also a low long-chain fat diet with MCT supplementation would be part of management[3]
See also
- Primary carnitine deficiency
- Carnitine palmitoyltransferase I deficiency
- Carnitine palmitoyltransferase II deficiency
References
- ↑ Reference, Genetics Home. "CACT deficiency". Genetics Home Reference. Archived from the original on 2017-02-28. Retrieved 2017-02-27.
- ↑ 2.0 2.1 Morales Corado, J. Andres; Lee, Chung U.; Enns, Gregory M. (1993). "Carnitine-Acylcarnitine Translocase Deficiency". GeneReviews®. University of Washington, Seattle. Archived from the original on 2023-05-29. Retrieved 2023-10-29.
- ↑ "GARD Rare Disease Information - Carnitine-acylcarnitine translocase deficiency - National Organization for Rare Disorders". rarediseases.org. 16 June 2022. Archived from the original on 15 August 2022. Retrieved 29 October 2023.
This article incorporates public domain text from The U.S. National Library of Medicine Archived 2019-02-04 at the Wayback Machine
External links
| Classification | |
|---|---|
| External resources |
- Pages with script errors
- All articles with unsourced statements
- Articles with unsourced statements from February 2017
- Articles with invalid date parameter in template
- Articles with unsourced statements from July 2017
- Articles with unsourced statements from September 2020
- Webarchive template wayback links
- Autosomal recessive disorders
- Rare diseases
- Fatty-acid metabolism disorders