Autoimmune hepatitis
| Autoimmune hepatitis | |
|---|---|
| Other names: Lupoid hepatitis | |
 | |
| Micrograph showing a lymphoplasmacytic interface hepatitis—the characteristic finding of autoimmune hepatitis. Liver biopsy. H&E stain. | |
| Specialty | Gastroenterology |
| Symptoms | None, tiredness, yellowish skin, abdominal pain[1] |
| Complications | Cirrhosis, liver failure, liver cancer[1] |
| Duration | Long-term[1] |
| Types | Type 1, type 2, variant[2] |
| Causes | Unknown[3] |
| Risk factors | Other autoimmune disease, genetics[3] |
| Diagnostic method | Based on symptoms, lab tests, and liver biopsy after ruling out other possible causes[3] |
| Differential diagnosis | Primary biliary cholangitis, primary sclerosing cholangitis, viral hepatitis[3] |
| Treatment | Prednisone, azathioprine[3] |
| Frequency | 11-25 per 100,000 (Europe)[3] |
Autoimmune hepatitis is a autoimmune disease in which the immune system attacks the liver and causes inflammation and liver damage.[1] Symptoms may vary from none, to abnormal liver enzymes, to tiredness, yellowish skin, and upper abdominal pain.[1][4] Complications may include cirrhosis, liver failure, and liver cancer.[1]
The cause is unknown.[3] Risk factors include other autoimmune diseases and genetics.[1][3] It is believed that an environmental event such as a virus or medication may trigger the disease in someone who is predisposed.[4] Diagnosis is based on symptoms, lab tests, and a liver biopsy after ruling out other possible causes.[3]
Treatment is often with a combination of prednisone and azathioprine for at least 3 years.[3] In those who develop liver failure, liver transplant may be an option.[3] Following a liver transplant about 33% have a recurrence.[3] Without treatment survival at 10 years is about 10%.[3]
In Europe about 11 to 25 per 100,000 people are affected with about 1 to 2 per 100,000 newly developing the disease each year.[3] Females are affected 3.6 times more often than males.[1][3] All ages may be affected.[5] It was first described in the 1950s by Waldenstrom.[2]
Signs and symptoms
Autoimmune hepatitis may present completely asymptomatic (12–35% of the cases), with signs of chronic liver disease, or acute or even fulminant hepatic failure.[6][7]
People usually present with one or more nonspecific symptoms, sometimes of long lasting duration, as fatigue, general ill health, lethargy, weight loss, mild right upper quadrant pain, malaise, anorexia, nausea, jaundice or joint pain (arthralgia), especially affecting the small joints. Rarely, rash or unexplained fever may appear. In women, the absence of menstruation (amenorrhoea) is a frequent feature. Physical examination may be normal, but it may also reveal signs and symptoms of chronic liver disease. Many people have only laboratory abnormalities as their initial presentation, as unexplained increase in transaminases and are diagnosed during an evaluation for other reasons. Others have already developed cirrhosis at diagnosis.[7] Of note, alkaline phosphatase and bilirubin are usually normal.
Autoimmune hepatitis frequently appears associated with other autoimmune conditions, mainly celiac disease, vasculitis, and autoimmune thyroiditis.[6]
Cause
About 60% of people have chronic hepatitis that may mimic viral hepatitis, but without serologic evidence of a viral infection. The disease is strongly associated with anti-smooth muscle autoantibodies. There is currently no conclusive evidence as to any specific cause.[8]
Diagnosis
The diagnosis of autoimmune hepatitis is best achieved with a combination of clinical, laboratory, and histological findings after excluding other etiological factors (e.g. viral [such as the Epstein-Barr virus], hereditary, metabolic, cholestatic, and drug-induced liver diseases). The requirement for histological examination necessitates a liver biopsy, typically performed with a needle by the percutaneous route, to provide liver tissue.
Autoantibodies
A number of specific antibodies found in the blood (antinuclear antibody (ANA), anti-smooth muscle antibody (SMA), anti-liver kidney microsomal antibodies (LKM-1, LKM-2, LKM-3), anti soluble liver antigen (SLA), liver–pancreas antigen (LP), and anti-mitochondrial antibody (AMA)) are of use, as is finding an increased immunoglobulin G level. The presence of anti-mitochondrial antibody is more suggestive of primary biliary cholangitis.[9]
Histology
Histological features supportive of a diagnosis of autoimmune hepatitis include:[10]
- A mixed inflammatory infiltrate centered on the portal tract
- The inflammatory infiltrate may breach the interface between the portal tract and liver parenchyma: so-called interface hepatitis
- The most numerous cell in the infiltrate is the CD4-positive T cell.
- Plasma cells may be present within the infiltrate. These are predominantly IgG-secreting.
- Eosinophils may be present within the infiltrate.
- Emperipolesis, where there is penetration of one cell through another, within the inflammatory infiltrate
- Varying degrees of necrosis of periportal hepatocytes.
- In more severe cases, necrosis may become confluent with necrotic bridges forming between central veins.
- Hepatocyte apoptosis manifest as acidophils or apoptotic bodies.
- Rosettes of regenerating hepatocytes.
- Any degree of fibrosis from none to advanced cirrhosis
- Biliary inflammation without destruction of biliary epithelial cells in a minority of cases.
-
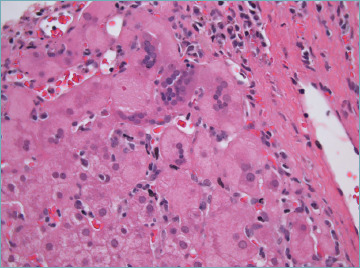
Giant cell transformation in autoimmune hepatitis
-
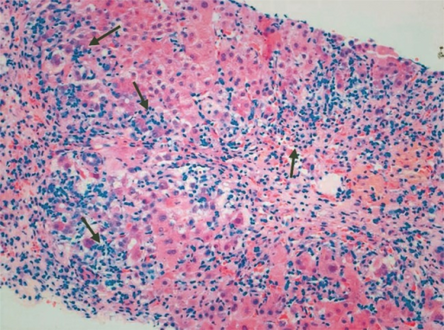
Portal and periportal inflammatory infiltrate- interface hepatitis (piecemeal necrosis) arrow(s)


Diagnostic scoring
Expert opinion has been summarized by the International Autoimmune Hepatitis Group, which has published criteria which utilize clinical, laboratory, and histological data that can be used to help determine if a patient has autoimmune hepatitis.[11] A calculator based on those criteria is available online.[12]
Overlap
Overlapping presentation with primary biliary cholangitis and primary sclerosing cholangitis has been observed.[13]
Classification
Four subtypes of autoimmune hepatitis are recognised, but the clinical utility of distinguishing subtypes is limited.
- Type 1 AIH. Positive ANA and SMA,[14] elevated immunoglobulin G (classic form, responds well to low dose steroids);
- Type 2 AIH. Positive LKM-1 (typically female children and teenagers; disease can be severe), LKM-2 or LKM-3;
- Type 3 AIH. Positive antibodies against soluble liver antigen[15] (this group behaves like group 1)[16] (anti-SLA, anti-LP)
- AIH with no autoantibodies detected (~20%)[citation needed] (of debatable validity/importance)
Treatment
Treatment may involve the prescription of immunosuppressive glucocorticoids such as prednisone, with or without azathioprine, and remission can be achieved in up to 60–80% of cases, although many will eventually experience a relapse.[17] Budesonide has been shown to be more effective in inducing remission than prednisone, and result in fewer adverse effects.[18] Those with autoimmune hepatitis who do not respond to glucocorticoids and azathioprine may be given other immunosuppressives like mycophenolate, ciclosporin, tacrolimus, methotrexate, etc. Liver transplantation may be required if patients do not respond to drug therapy or when patients present with fulminant liver failure.[19]
Prognosis
Autoimmune hepatitis is not a benign disease. Despite a good initial response to immunosuppression, recent studies suggest that the life expectancy of patients with autoimmune hepatitis is lower than that of the general population.[20][21][22] Additionally, presentation and response to therapy appears to differ according to race. For instance, African Americans appear to present with a more aggressive disease that is associated with worse outcomes.[23][24] If untreated, the mortality rate for severe autoimmune hepatitis may be as high as 40 percent.[5]
Outcomes with liver transplant are generally favorable, with a five-year survival greater than 80 percent.[5]
Epidemiology
Autoimmune hepatitis has an incidence of 1-2 per 100,000 per year, and a prevalence of 10-20/100,000. As with most other autoimmune diseases, it affects women much more often than men (70%).[25]
The disease may occur in any ethnic group and at any age, but is most often diagnosed in patients between age 40 and 50.[5]
In the US the 5-year prevalence rate was found to be 31 per 100,000 persons.[26]
History
Autoimmune hepatitis was previously called "lupoid" hepatitis. It was originally described in the early 1950s.[27]
Most patients do have an associated autoimmune disorder such as systemic lupus erythematosus. Thus, its name was previously lupoid hepatitis.
Because the disease has multiple different forms, and is not always associated with systemic lupus erythematosus, lupoid hepatitis is no longer used. The current name at present is autoimmune hepatitis (AIH).
References
- ↑ 1.0 1.1 1.2 1.3 1.4 1.5 1.6 1.7 "Definition & Facts for Autoimmune Hepatitis | NIDDK". National Institute of Diabetes and Digestive and Kidney Diseases. Archived from the original on 4 February 2021. Retrieved 13 February 2021.
- ↑ 2.0 2.1 "Autoimmune Hepatitis". NORD (National Organization for Rare Disorders). Archived from the original on 10 March 2021. Retrieved 14 February 2021.
- ↑ 3.00 3.01 3.02 3.03 3.04 3.05 3.06 3.07 3.08 3.09 3.10 3.11 3.12 3.13 3.14 Linzay, CD; Sharma, B; Pandit, S (January 2020). "Autoimmune Hepatitis". PMID 29083819.
{{cite journal}}: Cite journal requires|journal=(help) - ↑ 4.0 4.1 "Symptoms & Causes of Autoimmune Hepatitis | NIDDK". National Institute of Diabetes and Digestive and Kidney Diseases. Archived from the original on 2 March 2021. Retrieved 14 February 2021.
- ↑ 5.0 5.1 5.2 5.3 Manns, MP; Czaja, AJ; Gorham, JD; Krawitt, EL; Mieli-Vergani, G; Vergani, D; Vierling, JM; American Association for the Study of Liver, Diseases (June 2010). "Diagnosis and management of autoimmune hepatitis". Hepatology. 51 (6): 2193–213. doi:10.1002/hep.23584. PMID 20513004. S2CID 30356212.
- ↑ 6.0 6.1 Than NN, Jeffery HC, Oo YH (2016). "Autoimmune Hepatitis: Progress from Global Immunosuppression to Personalised Regulatory T Cell Therapy". Can J Gastroenterol Hepatol (Review). 2016: 1–12. doi:10.1155/2016/7181685. PMC 4904688. PMID 27446862.
- ↑ 7.0 7.1 Zachou K, Muratori P, Koukoulis GK, Granito A, Gatselis N, Fabbri A, et al. (2013). "Review article: autoimmune hepatitis -- current management and challenges". Aliment Pharmacol Ther (Review). 38 (8): 887–913. doi:10.1111/apt.12470. PMID 24010812. S2CID 29295458.

- ↑ "Autoimmune hepatitis | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 2018-04-18. Retrieved 2018-04-17.
- ↑ Alvarez, F; Berg, PA; Bianchi, FB; Bianchi, L; Burroughs, AK; Cancado, EL; Chapman, RW; Cooksley, WG; Czaja, AJ; Desmet, VJ; Donaldson, PT; Eddleston, AL; Fainboim, L; Heathcote, J; Homberg, JC; Hoofnagle, JH; Kakumu, S; Krawitt, EL; Mackay, IR; MacSween, RN; Maddrey, WC; Manns, MP; McFarlane, IG; Meyer zum Büschenfelde, KH; Zeniya, M (November 1999). "International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis". Journal of Hepatology. 31 (5): 929–38. doi:10.1016/S0168-8278(99)80297-9. PMID 10580593.
- ↑ Webb, GJ; Hirschfield, GM; Krawitt, EL; Gershwin, ME (24 January 2018). "Cellular and Molecular Mechanisms of Autoimmune Hepatitis". Annual Review of Pathology. 13: 247–292. doi:10.1146/annurev-pathol-020117-043534. PMID 29140756.
- ↑ Alvarez F, Berg PA, Bianchi FB, et al. (November 1999). "International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis". J. Hepatol. 31 (5): 929–38. doi:10.1016/S0168-8278(99)80297-9. PMID 10580593.
- ↑ "Autoimmune Hepatitis Calculator". Archived from the original on 2017-10-27. Retrieved 2008-05-09.
- ↑ Washington MK (February 2007). "Autoimmune liver disease: overlap and outliers". Mod. Pathol. 20 Suppl 1: S15–30. doi:10.1038/modpathol.3800684. PMID 17486048.
- ↑ Bogdanos DP, Invernizzi P, Mackay IR, Vergani D (June 2008). "Autoimmune liver serology: Current diagnostic and clinical challenges". World J. Gastroenterol. 14 (21): 3374–3387. doi:10.3748/wjg.14.3374. PMC 2716592. PMID 18528935. Archived from the original on 2016-05-28. Retrieved 2008-07-05.
- ↑ "autoimmune hepatitis". Archived from the original on 2012-07-01. Retrieved 2008-07-05.
- ↑ "Medscape & eMedicine Log In". Archived from the original on 2012-04-03. Retrieved 2008-07-05.
- ↑ Krawitt EL (January 1994). "Autoimmune hepatitis: classification, heterogeneity, and treatment". Am. J. Med. 96 (1A): 23S–26S. doi:10.1016/0002-9343(94)90186-4. PMID 8109584.
- ↑ Manns, MP; Strassburg, CP (2011). "Therapeutic strategies for autoimmune hepatitis". Digestive Diseases. 29 (4): 411–5. doi:10.1159/000329805. PMID 21894012. S2CID 42014343.
- ↑ Stephen J Mcphee, Maxine A Papadakis. Current medical diagnosis and treatment 2009 page.596
- ↑ Hoeroldt, Barbara; McFarlane, Elaine; Dube, Asha; Basumani, Pandurangan; Karajeh, Mohammed; Campbell, Michael J.; Gleeson, Dermot (2011). "Long-term Outcomes of Patients With Autoimmune Hepatitis Managed at a Nontransplant Center". Gastroenterology. 140 (7): 1980–1989. doi:10.1053/j.gastro.2011.02.065. ISSN 0016-5085. PMID 21396370.
- ↑ Ngu, Jing Hieng; Gearry, Richard Blair; Frampton, Chris Miles; Stedman, Catherine A.M. (2013). "Predictors of poor outcome in patients with autoimmune hepatitis: A population-based study". Hepatology. 57 (6): 2399–2406. doi:10.1002/hep.26290. ISSN 0270-9139. PMID 23359353. S2CID 8890549.
- ↑ Ngu, Jing Hieng; Gearry, Richard Blair; Frampton, Chris Miles; Malcolm Stedman, Catherine Ann (2012). "Mortality and the risk of malignancy in autoimmune liver diseases: A population-based study in Canterbury, New Zealand". Hepatology. 55 (2): 522–529. doi:10.1002/hep.24743. ISSN 0270-9139. PMID 21994151. S2CID 30580281.
- ↑ Lim, Kie N.; Casanova, Roberto L.; Boyer, Thomas D.; Bruno, Christine Janes (2001). "Autoimmune hepatitis in African Americans: presenting features and response to therapy". The American Journal of Gastroenterology. 96 (12): 3390–3394. ISSN 0002-9270. PMID 11774954.
- ↑ Verma, Sumita; Torbenson, Michael; Thuluvath, Paul J. (2007). "The impact of ethnicity on the natural history of autoimmune hepatitis". Hepatology. 46 (6): 1828–1835. doi:10.1002/hep.21884. ISSN 0270-9139. PMID 17705297. S2CID 12506294.
- ↑ "Autoimmune Hepatitis". Archived from the original on 2008-05-09. Retrieved 2008-05-09.
- ↑ Tunio, Nahel A.; Mansoor, Emad; Sheriff, Mohammed Z.; Cooper, Gregory S.; Sclair, Seth N.; Cohen, Stanley M. (November 2021). "Epidemiology of Autoimmune Hepatitis (AIH) in the United States Between 2014 and 2019: A Population-based National Study". Journal of Clinical Gastroenterology. 55 (10): 903–910. doi:10.1097/MCG.0000000000001449. Archived from the original on 13 April 2023. Retrieved 13 April 2023.
- ↑ Aizawa Y, Hokari A (2017). "Autoimmune hepatitis: current challenges and future prospects". Clin Exp Gastroenterol (Review). 10: 9–18. doi:10.2147/CEG.S101440. PMC 5261603. PMID 28176894.
External links
| Classification | |
|---|---|
| External resources |